Stand: 15.12.2007
![]()
|
Stand: 15.12.2007 |
|
|
|
150 Jahre Elementanalytik im Hessischen Landwirtschaftlichen
Untersuchungswesen
(R.Ellinghaus, Festschrift '150 Jahre
Lanwirtschaftliches Untersuchungs- und Forschungswesen in Hessen' 10/2007,
ISBN: 3-9806860-9-4)
Begriffe
Überschriften sollen kurz und doch umfassend sein, ein
meist unerfüllbares Anliegen. Der Begriff ‚Elementanalytik’ ist eher der
modernen Instrumentellen Analytik entlehnt, ist er auch der richtige, das
anorganisch-analytische Schaffen im Landwirtschaftlichen Untersuchungswesen von
anderthalb Jahrhunderten abzubilden?
„Nun, sicher“, sagt der nicht spezialisierte
Anorganiker, „im Fokus der Anorganischen Chemie stehen schließlich die
chemischen Elemente.“
„Langsam“, sagt der Analytiker aus dem modernen
elementanalytischen Labor, „Elementanalytik ist für mich
atomspektrometrisches Messen. Bei den klassischen Methoden waren chemische
Verbindungen die Analyten, das nenne ich eher Molekül- als Elementanalytik.“
„Ich bin mir nicht sicher“, sagt ein Historiker,
„früher war doch eher von Nähr- und Mineralstoff- statt Elementanalytik die
Rede, und heute geht es doch meist um Schwermetalle, Spuren oder
Kontaminanten.“
„Aber“, sagt ein anderer Historiker, „haben nicht
bedeutende Chemiker bestimmte anorganische Analysenverfahren schon vor mehr als
150 Jahren Elementaranalysen genannt, ohne, dass sie bis heute jemand korrigiert
hätte?“
Recht haben sie aus ihrem jeweiligen Blickwinkel alle. Da
das so ist, ist ‚Elementanalytik’ auch ein geeigneter Oberbegriff. Die
kleine fiktive Diskussion soll in der Folge helfen, die wissenschaftlichen, aber
auch politisch-gesellschaftlichen Felder, die von der
anorganisch-landwirtschaftlichen Analytik tangiert werden, als zusammengehörig
und als deren Facetten zu verstehen.
Wegbereiter
Die Analytische Chemie mit ihrer Frage nach der
qualitativen und quantitativen Zusammensetzung von Stoffen ist die Disziplin,
die die Chemie aus der Alchemie heraus begründete und zu einer Wissenschaft
machte. Die Agrikulturchemie und hier im wesentlichen die anorganische
Agrikulturchemie ist die Disziplin, die die Analytische Chemie erstmals zur Lösung
eines großen gesellschaftlichen Problems befähigte, der bedrohlichen Ernährungssituation
im 19. Jahrhundert.
Der wissenschaftliche Ruhm wurde dabei zunächst nur
Justus von Liebig (1803-1873) (Abb.1) zuteil, er galt über ein Jahrhundert als
alleiniger Begründer der Agrikulturchemie und der Lehre der Mineralstoffernährung
der Pflanzen. Heute kennt der Chemiker im landwirtschaftlichen Untersuchungs-
und Forschungswesen auch Carl Sprengel (1787-1859) (Abb.2), der über ein
Jahrzehnt vor Liebig publizierte und dabei auch schon das Minimumgesetz
postulierte, nach dem derjenige Nährstoff unter mehreren das Pflanzenwachstum
begrenzt, der vom Bedarf der Pflanze her zuerst zur Neige geht.
Beide Wissenschaftler teilen sich nun die Ehre, der
Mineraldüngung mit ihren epochalen Auswirkungen auf die Ernährung der Weltbevölkerung
zum Durchbruch verholfen zu haben.
Als Element mit dem größten Düngebedarf erwies sich
Phosphor (P). Pflanzen nehmen es aus dem Boden anionisch in Form von gelösten
Phosphaten, den Salzen der Phosphorsäure, auf. Unter Agrarwissenschaftlern bürgerte
es sich ein, deshalb von einem Mangel an Phosphorsäure zu sprechen, obwohl die
Phosphormineralien, mit denen gedüngt wurde, in ihrer chemischen Struktur sehr
wohl als Phosphate bekannt waren und auch so bezeichnet wurden. Noch heute ist
es traditionell üblich, den wie immer auch bestimmten Gehalt an Phosphor in Düngern
und Böden auf das Anhydrid der Phosphorsäure, P2O5
(di-Phosphorpentoxid) zu beziehen.
Der Mangel an Phosphor bewirkte nach dem Minimumprinzip,
dass auch andere Nährstoffelemente, allen voran Stickstoff (N) und Kalium (K)
nicht hinreichend von den Pflanzen ausgenutzt werden konnten, sofern ein Angebot
von seiten der Böden überhaupt bestand. Pflanzen nehmen Stickstoff in Form
seiner gelösten Salze anionisch über Nitrate (Salze der Salpetersäure) u.a.
oder kationisch über Ammoniumsalze (Salze des Ammoniaks) auf. Daneben sind
bestimmte Pflanzen (Leguminosen) auch in der Lage, Stickstoff über einen
bakteriellen Prozess direkt aus der Luft zu assimilieren. Als Bezugsgröße
etablierte sich in Düngern der elementare N-Gehalt. Kalium wird von Pflanzen
kationisch über gelöste Kaliumsalze aufgenommen. Als Bezugsgröße wurde hier
analog dem Phosphor mit K2O (di-Kaliumoxid, Kurzbezeichnung: Kali)
ein Anhydrid gewählt, das der Kalilauge.
|
|
|
|
Abb.1 Justus von Liebig |
Abb.2 Carl Sprengel |
Liebig’s Gesamtwerk ist für den Analytiker natürlich
ungleich interessanter als das Sprengel’s und nicht auf die Agrikulturchemie
beschränkt. Er war der Chemiker und Analytiker, der sich der Agrarwissenschaft
zuwandte, Sprengel kam von der Agrarwissenschaft und bediente sich
naturwissenschaftlicher Erkenntnisse. Mit ein wenig Lokalkolorit darf auch
angemerkt sein, dass Liebig Hesse war und rund 30 Jahre in Hessen
wissenschaftlich wirkte und lehrte, womit ihm neben seinen zahlreichen anderen
Ehrungen gut und gerne auch der Status eines Ehrenmitarbeiters im Hessischen
Landwirtschaftlichen Untersuchungswesen zustände. Die Gründung der
Landwirtschaftlichen Versuchsstationen in Hessen erlebte er allerdings nur von
fern als ‚Deserteur’ in München.
Liebig war als glänzender Experimentator bekannt und
erfand zahlreiches Laborgerät. So gilt er zusammen mit Lavoisier (1743-1794)
und Berzelius (1779-1848) auch als Wegbereiter der Elementaranalyse. Eine seiner
Erfindungen war der unten abgebildete Fünf-Kugel- oder Kali-Apparat (Abb.3), in
dem zur quantitativen Bestimmung des Elements Kohlenstoff (C) in einer
Verbindung das durch Verbrennung der Verbindung aus Kohlenstoff gebildete
Kohlendioxid (CO2) in Kalilauge (KOH) absorbiert werden konnte. Die
Gewichtszunahme durch das sich bildende Kaliumcarbonat (K2CO3)
wurde ausgewogen und erlaubte auf Grund der bekannten Zusammensetzung des
Carbonats die Berechnung der Menge des absorbierten Kohlendioxids und schließlich
des Kohlenstoffanteils in der Verbindung.
Über Verbrennungsanalytik wird Kohlenstoff in
landwirtschaftlichen Produktionsmitteln und Produkten auch heute noch bestimmt,
nur das Messprinzip hat sich von einer Wägungsmethodik (Gravimetrie) zu einer
später noch vorzustellenden physikalischen Messmethodik gewandelt.
Die klassische wie moderne Elementaranalyse als Teil der
Elementanalytik ist beschränkt auf nichtmetallische Elemente (Abb.6), deren
Verbindungen nach Oxidation, Reduktion oder Säure/Base-Reaktion gasförmige
Reaktionsprodukte bilden, die sich entweder in Flüssigkeiten absorbieren und
darin gravimetrisch, titrimetrisch o.a. bestimmen lassen oder einer Bestimmung
in der Gasphase zugänglich sind.
Ein Verfahren besonderer Bedeutung hinsichtlich seines
breitgefächerten Einsatzes in der landwirtschaftlichen Elementaranalyse damals
wie heute stellt das Kjeldahl-Verfahren zur Bestimmung von Stickstoff dar.
Benannt nach seinem Erfinder Johan Kjeldahl (1849-1900) basiert es auf der
chemischen Umsetzung aller Stickstoffverbindungen einer Probe zu Ammoniumsalzen
(NH4X), die bei Zugabe von Natronlauge Ammoniak (NH3)
freisetzen, der destillativ (Abb.4) ausgetrieben, in einer schwachen Säure
absorbiert und darin titrimetrisch bestimmt werden kann. Über die Menge an
Ammoniak kann der Stickstoffgehalt der Probe berechnet werden.
|
|
|
|
Abb.3 Kali-Apparat |
Abb.4 Kjehldahl-Destille |
Das Kjeldahl-Verfahren erblickte 1883 das Licht der Welt. Gehen wir nun aber zunächst wieder 30 Jahre zurück, um bei der Gründung der hessischen Landwirtschaftlichen Versuchstationen dabei zu sein, und um deren anorganisch-analytisches Schaffen zu verfolgen, in dem es auch immer wieder um die Adaption neuer Verfahren und Ideen wie die eines Kjeldahls gehen wird.
Der Beginn
Chemie und Landwirtschaft waren sich bei Liebig und
Sprengel begegnet. Nun bestand Bedarf an der Vermittlung der neuen Erkenntnisse
an den Praktiker in der Landwirtschaft und am Ausbau des Wissens durch
Angewandte Forschung in Zusammenarbeit mit der Praxis. Von Landwirtschaftlichen
Versuchsstationen wurde dieses Potential erwartet, und sie wurden ab 1850 überall
in Deutschland von landwirtschaftlichen Organisationen und Verbänden gegründet,
so auch im Kurfürstentum Hessen-Kassel 1857 und im Großherzogtum
Hessen-Darmstadt 1869.
Vermittlung von Erkenntnissen geht einher mit Vertrauen
schaffen in die Erkenntnisse, d.h. seriöse Beratung bedurfte der Unterstützung
verlässlicher Untersuchungsergebnisse. Sie halfen einerseits dem
wissenschaftlichen Landwirtschaftlichen Versuchswesen mit ergänzenden Daten,
zum anderen aber dem Praktiker bei der Kontrolle, ob seine erworbenen
landwirtschaftlichen Produktionsmittel auch den Deklarationen entsprachen.
Zu Anfang bestimmten eindeutig die Forschungstätigkeiten
das Geschehen in den hessischen Stationen. Die Kontrolltätigkeiten weiteten
sich dann aber ab den 80er-Jahren soweit aus, dass um die Jahrhundertwende
Stimmen laut wurden, die forderten, beides institutionell zu entkoppeln, um der
Forschung wieder breiteren Raum zu verschaffen. Aus heutiger Sicht mit dem
Wissen, wie positiv sich Angewandte Forschung und Praxis schließlich
beeinflussten, ist zu begrüßen, dass dies noch 100 Jahre dauern sollte.
Das erste halbe Jahrhundert
Der anorganisch-chemischen Analytik war es egal, wem sie
diente, aber natürlich nahm ihre Bedeutung mit der Ausweitung der Kontrolltätigkeiten
zu. Die landwirtschaftlichen Produktionsmittel, um die sie sich zunächst kümmern
musste, waren die mineralischen und organisch-mineralischen Düngemittel mit den
Nährelementen, die die obere Reihe der Abb.5 zeigt, und die pflanzlichen
Futtermittel, bei denen sich die Analytk ganz auf den Stickstoff als Basisanalyt
für die Berechnung des Rohproteins fokussierte. Rohprotein definiert die Summe
aller stickstoffhaltigen Nährstoffe in organischen Futtermitteln (Eiweiße,
Amide).
|
|
|
Abb.5 Pflanzennährstoffe |
Zum Einsatz kamen schließlich die quantitativen
Analysenverfahren, die heute ‚klassisch’ genannt werden: Gravimetrie (Messgröße
(MG): Gewicht), Titrimetrie (Synonyme: Volumetrie, Maßanalyse) (MG: Volumen
einer Flüssigkeit) und Gasvolumetrie (MG: Volumen eines Gases). Indes war die
quantitative analytische Chemie bis zur Mitte des 19.Jahrhunderts kaum über die
ersten Anfänge hinaus, und die z.T. aufwändigen Methoden eigneten sich zwar für
rein wissenschaftliche Zwecke, nicht aber für die Serienanalytik. So
konzentrierte sich in den ersten Jahrzehnten die Hauptarbeit der Analytiker auf
die Entwicklung serientauglicher Bestimmungsmethoden. 1888 gab dann die Gründung
des ‚Verbandes landwirtschaftlicher Versuchstationen im Deutschen Reiche’,
dem heutigen ‚Verband der Deutschen Landwirtschaftlichen Untersuchungs- und
Forschungsanstalten (VDLUFA)’ einen ganz entscheidenden synergetischen Schub für
die Verbesserung und Validierung der schon in der Praxis befindlichen und die
Entwicklung neuer Methoden.
Im Fokus der Düngemittelanalytik stand jahrzehntelang
der Phosphor. Dabei war bedingt durch die Erkenntnis, dass es unterschiedlich
schnell pflanzenverfügbare Phosphatbindungsformen gibt, auch eine
differenzierte Extraktionschemie zu entwickeln und nicht nur die Frage nach dem
P-Gesamtgehalt zu stellen.
In Darmstadt wurde unter der Leitung des über die
Grenzen bekannten Düngungsexperten Paul Wagner (1843-1930) ein Verfahren der Fällung
von citronensäurelöslichem Phosphat zur Reife entwickelt. Hierbei wurde zunächst
durch Zugabe von Ammoniak (NH3) die Fällung schwerlöslicher
Phosphate, z.B. des Calciums oder Eisens verhindert und dann mit
Magnesiumchlorid (MgCl2) quantitativ das komplexe Doppelsalz
Magnesiumammoniumphosphat (MgNH4PO4) gefällt, isoliert,
zu Magnesiumpyrophosphat (Mg2P2O7) geglüht und
in dieser oxidischen Form gravimetrisch bestimmt. Das Doppelsalz konnte und kann
vice versa natürlich auch für eine quantitative Bestimmung von Magnesium (Mg)
genutzt werden.
Auch in der kurhessischen Station, zu dieser Zeit in
Marburg, wurde mit Magnesiumammoniumphosphat experimentiert und eine
titrimetrische Methode entwickelt. Das Salz wurde mit überschüssiger 0,2
molarer Salzsäure umgesetzt und die nicht verbrauchte Salzsäure mit 0,2
molarer Natronlauge zurücktitriert. Als Indikator diente der Farbstoff
Karminrot, der den Neutralisationspunkt von Säure und Lauge durch einen
Farbumschlag von gelbbraun nach violett anzeigte. Aus der Reaktionsgleichung der
Umsetzung Salz mit Säure und der nach der Titration bekannten Menge der für
die Umsetzung verbrauchten Säure konnte die Menge Magnesiumammoniumphosphat und
daraus der Phosphorgehalt berechnet werden.
Die Stickstoffbestimmung in Futtermitteln wurde bis zur
Einführung der Kjeldahl-Methode nach Vorgaben von Jean Baptiste Dumas
(1800-1884) durchgeführt. Dumas’ Name steht auch heute noch für die
quantitative verbrennungsanalytische Bestimmung von Nichtmetallelementen (Abb.6)
und ziert so manche gerade veröffentlichte internationale Norm. Moderne
Anwendungen kommen später noch zur Sprache, beim auf dem Urverfahren von 1840
basierenden Ablauf wurde das Futtermittel zusammen mit Kupferoxid (CuO/CuO2)
in einem geschlossenen System bei hohen Temperaturen verbrannt, entstehende
Feuchtigkeit (per Trocknungsmittel) und entstehendes Kohlendioxid (z.B. per
Kali-Apparat) aus den Verbrennungsgasen absorbiert und der allein verbleibende
Stickstoff über sein Gasvolumen bestimmt.
|
|
|
Abb.6 Metalle und Nichtmetalle unter den Elementen |
Das Kupferoxid ist Sauerstofflieferant und wird dabei zu
Kupfer (Cu) reduziert, welches seinerseits die bei der Verbrennung entstandenen
Stickoxide (NO, NO2 u.a.) zu Stickstoff reduziert und selbst wieder
zu Kupferoxid oxidiert wird. Da das Kupferoxid am Ende unverändert vorliegt,
erfüllt es die Definition eines Katalysators.Somit ist Dumas auch ein Pionier
der Katalyse. Was er wohl sagen würde, wenn er wüsste, dass heutige Forscher
nach wie vor auf der Suche nach geeigneten Katalysatoren für
Verbrennungsprozesse sind? Deren Ziele heißen allerdings Abgasreinigung und
Umweltschutz.
Bis 1890 wurden andere Probenmatrices als Düngemittel
und Futtermittel elementaranalytisch nur im Rahmen von Forschungen untersucht.
1868 gab es aus der Marburger Station z.B. zwei Hauspublikationen zu
Pflanzenaschen und Pflanzenfetten. In den 90er-Jahren beschäftigte man sich
dann erstmalig mit Bodenanalytik.
An einer systematischen Bodenuntersuchung wie heute zum
Zwecke eines optimierten Düngereinsatzes bestand damals allerdings noch kein
Interesse. Eine Bodenuntersuchung galt als zu teuer für den Landwirt, man
verließ sich bei der Beratung auf die vorhandene Erfahrung aus dem
Landwirtschaftlichen Versuchswesen.
Eine aus den ältesten noch verfügbaren Jahresberichten
der Marburger Station von 1895 bis zur Jahrhundertwende zusammengestellte
Tabelle (Abb.7) zeigt den schon beachtlichen Untersuchungsumfang, die steigenden
Untersuchungszahlen und die für diese Zeit typische Verteilung auf die
verschiedenen Matrices. Düngemittel und Futtermittel dominierten wie schon die
beiden Jahrzehnte zuvor, neu erschienen Böden und Wässer, meist Trinkwässer
und Mineralwässer, aber auch Abwässer.
Obwohl - das scheint zeitlos für Jahresberichte
unabwendbar zu sein - die Chronisten mit dem Zählen der Proben zumeist ihre
Pflicht für erfüllt hielten und zu Analysenverfahren schwiegen, kann dem
anorganisch-analytischen Schaffen doch sicher jede Düngemittel-, Futtermittel-
und Bodenprobe mit einer oder mehreren Analysen zugerechnet werden. Wässer
hingegen wurden überwiegend sensorisch und bakteriologisch bewertet und nur bei
bestimmten Fragestellungen elementanalytisch untersucht.
|
Probenart |
1895 |
1896 |
1897 |
1898 |
1899 |
1900 |
|
Düngemittel |
717 |
949 |
1306 |
1542 |
1602 |
1712 |
|
Futtermittel |
314 |
345 |
348 |
416 |
504 |
517 |
|
Boden |
10 |
7 |
11 |
21 |
55 |
83 |
|
Wasser |
320 |
380 |
278 |
311 |
255 |
411 |
Abb.7 Untersuchte Proben in Marburg 1895-1900
In der Darmstädter Station war das Probenaufkommen ähnlich.
Auf Grund der bedeutenden Stellung der Düngeforschung dort gab es aber eine
noch größere Präferenz der Düngemitteluntersuchungen als in Marburg. 1885
konnten durch einen Standortwechsel innnerhalb Darmstadts neue modernere
Laboratorien bezogen und die Untersuchungskapazitäten ausgebaut werden. Werfen
wir einmal einen Blick in ein typisches anorganisch-chemisches Laboratorium um
1900. Abb.8 zeigt uns das Darmstädter Düngemittellabor.
|
|
|
Abb.8 Düngemittellabor in Darmstadt um 1900 |
Von den 717 Marburger Düngerproben des Jahres 1895 wurde
bei 573 der Phosphor-, bei 77 der Phosphor- und Stickstoff-, bei 24 der
Stickstoff-, bei 20 der Calcium-, bei 15 der Kalium- und bei 8 Proben ein
anderer Elementgehalt bestimmt. Dieses Muster der Bedeutung der einzelnen Nährstoffelemente
bei den Düngerkontrolluntersuchungen blieb bis zum 1. Weltkrieg ziemlich unverändert.
Von den 1712 Düngerproben im Jahre 1900 erfuhren 1321
eine Phosphorbestimmung. In diesem Jahr ist erstmals auch etwas über die
Anteile unterschiedlicher Probenvorbereitung zu lesen: 1067 Proben wurden mit Säure
aufgeschlossen oder mit Citronensäure extrahiert, bei 248 Proben wurde der
Phosphorgehalt aus einer wässrigen Extraktion bestimmt.
Der Jahresbericht von 1900 ist für den Elementanalytiker
auf historischer Spurensuche aber besonders wegen erstaunlicher Mitteilungen aus
der Wasseranalytik eine aufregende Fundgrube. In 7 Trinkwässern, ist zu lesen,
sei der Blei- (Pb) und in 5 Mineralwässern der Kupfergehalt (Cu) beanstandet
worden. Und für die Bleianalysen werden auch Messergebnisse angeboten: 0,2 bis
1 Milligramm Blei pro Liter (mg/l). Es muss wohl eine gewisse Überheblichkeit
des heute mit solch niedrigen Gehalten jeden Tag hantierenden Chronisten sein,
dass er ungläubig das Datum prüfte: doch es blieb das Jahr 1900. Dass überhaupt,
und nun muss der Begriff früher erscheinen als eigentlich geplant, bereits
‚Spurenanalytik’ betrieben wurde, verblüffte schon genug, die dabei
erreichbare Nachweisstärke für ein offensichtliches Routineverfahren war aber
gänzlich überraschend. So stieß der ‚Spurenanalytiker’ von heute bei
seiner historischen Spurensuche unversehens früh auf ‚Spurenelemente’.
Spannend, da leider nicht überliefert, ist die Frage:
Welches Messverfahren wurde hier eingesetzt? Die klassischen Verfahren
Gravimetrie und Titrimetrie scheinen für die Routine bei so niedrigen Gehalten
ungeeignet. Dass es Routineverfahren waren, legt einerseits die relativ
unkomplizierte Matrix Wasser nahe, andererseits lässt die Aussage, die Proben
seien beanstandet worden, den Schluss zu, sie stammten aus einer größeren
Serie. Drittens ist auch in den nachfolgenden Jahren mehrfach nachzulesen, dass
Mineralwässer wegen erhöhter Blei- und Kupfergehalte auffällig geworden
seien, wohl also ein Untersuchungsverfahren, das regelmäßig zum Einsatz kam,
existiert haben muss. Die Angabe von Gehalten blieb allerdings dem Jahresbericht
von 1900 vorbehalten und wurde nicht wieder bestätigt.
Die Gravimetrie als Messmethode auszuschließen, ist
allein durch die Vorstellung vom Aufwand der Isolierung und Reinigung von
geringsten Mengen ausgefällter Blei- und Kupfersalze her zu begründen, die
Waagentechnik (Abb.9) war durchaus schon soweit entwickelt, bis 0,1 Milligramm
wiegen zu können. Die Titrimetrie damaligen Entwicklungsstandes, die noch keine
komplexometrischen Reagenzien für Metalle und keine automatisierten
Mikromethoden kannte, ist indes gewiß nicht die in Frage kommende Methode.
|
|
|
Abb.9 Analysenwaage um 1900 |
Viel wahrscheinlicher ist ein kolorimetrisches oder
elektrochemisches Verfahren. Die Kolorimetrie (siehe nächstes Kapitel) war wie
die Titrimetrie allerdings auf ein gutes menschliches Auge angewiesen, das
kleinste Farbnuancen zu erkennen vermochte. Es müssten schon intensivste Anfärbungen
von Blei- und Kupferverbindungen bekannt gewesen sein, um in dem niedrigen
Konzentrationsbereich arbeiten zu können. Favorit auf der Spekulationsskala wäre
demnach eine elektrochemische Methode.
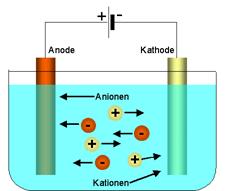
Nachdem bereits in den 30er-Jahren des 19.Jahrhunderts
Michael Faraday (1791-1867) seine bahnbrechenden Arbeiten zur Elektrolyse
(Abb.10) mit den nach ihm benannten Gesetzen vorgestellt hatte, war die
quantitative elektrochemische Analytik als instrumentelles Analysenverfahren um
1900 bereits deutlich weiter entwickelt als die heute dominierende quantitative
optische bzw. spektrometrische Analytik (siehe nächstes Kapitel). Für Blei-
und Kupferbestimmungen eignen sich die Elektrogravimetrie und die Coulometrie.
In beiden Fällen wird die Gesamtmenge des Analyten an einer Elektrode reduktiv
oder oxidativ abgeschieden, wenn eine für den Analyt spezifische konstante
Spannung an die Elektrolysezelle gelegt wird. Kupfer scheidet sich dabei
elementar kathodisch ab, Blei als Bleioxid (PbO2) anodisch. Bei der
Elektrogravimetrie wird der Analytgehalt aus der Gewichtsdifferenz der Elektrode
vor und nach der Elektrolyse ermittelt, bei der Coulometrie aus der zwischen den
Elektroden fließenden Ladungsmenge während des Abscheideprozesses, die sich
zur Menge des Analyten proportional verhält.
|
|
|
Abb.10 Schema einer Elektrolyse |
In der heutigen Trinkwasserverordnung finden wir einen
Grenzwert von 0,025 mg/l für Blei. Es war also eine Menge Blei in den
beanstandeten Wässern, das aus bleiernen Wasserleitungen stammte. Ein Problem
ist das in alten Häusern bisweilen heute noch. Auch die Kupferbelastung der
Mineralwässer ist nachvollziehbar. Die damals gebräuchlichen verzinnten
Kupferleitungen der Produktionsstätten hatten ihre Oberflächenversiegelung
verloren.
Um die Jahrhundertwende und im ersten Jahrzehnt danach
taucht noch ab und an ein Exot im Elementespektrum des Untersuchungswesens in
Marburg auf. Mal wird Zink in getrockneten Äpfeln mit Gehalten um 0,2%, mal
Blei in Zinndeckeln von Trinkkrügen mit Gehalten um 20% und mal Blei in Tonkrügen
ohne überlieferte Gehaltsangaben analysiert und erwähnt, doch kann hier von
‚Spurenanalytik’ keine Rede sein. In dieser Hinsicht stehen die
Wasserergebnisse von 1900 ziemlich einmalig da. Aber es gibt noch einen anderen
Sammelbegriff für die vorstehenden Elemente, der hier eingeführt gehört und
später noch genauer zu definieren sein wird: ‚Schwermetalle’.
Leider sind aus dieser Zeit keine Detailberichte aus
Darmstadt auffindbar, um vielleicht auch dort etwas von Exoten zu entdecken.
Obwohl die beiden hessischen Stationen unter ihren jeweiligen Obrigkeiten strikt
getrennt arbeiteten, waren andererseits die wissenschaftlichen Erkenntnisse und
elementanalytischen Methoden der Zeit Allgemeingut und letzlich beide Häuser
auch im ‚Verband der Landwirtschaftlichen Versuchsstationen’ tätig, woraus
mit hoher Wahrscheinlichkeit auch für Darmstadt auf die ersten Schwermetall-
und vielleicht ersten Spurenanalysen geschlossen werden kann.
Beschließen wir die ersten rund 50 Jahre
anorganisch-analytischen Schaffens zum Wohle der Landwirtschaft in Hessen mit
der Nachricht von neuen Laboratorien auch für die kurhessische Station. Die
bekam sie, als sie 1910 von Marburg an ihren heutigen Standort in
Kassel-Harleshausen verlegt wurde.
![]()
Licht im Dunkel der Elementanalytik
Ohne optische Messverfahren geht heute wenig in der
Elementanalytik. Bevor das zweite halbe Jahrhundert, wo sie Einzug in das
Landwirtschaftliche Untersuchungswesen hielten, und das dritte, wo sie sich
unentbehrlich machten, an uns vorüberziehen kann, sollen ein paar Grundbegriffe
helfen, die verschiedenen Varianten der optischen Verfahren im Bezug zueinander
kennenzulernen.
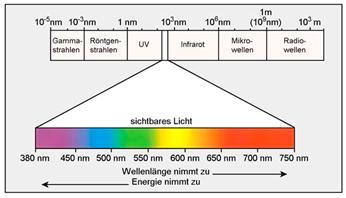
All das, was nun Revue passieren soll, betrifft die
Wechelwirkung von Materie mit Licht oder richtiger: elektromagnetischer
Strahlung (Abb.11). Deren Strahlungsenergie kann von Materie in Portionen
absorbiert oder emittiert werden. Wenn Materie Strahlung absorbiert, verändert
sich in ihr etwas von einem definierten energieärmeren Zustand A zu einem
definierten energiereicheren Zustand B, der ‚angeregt’ genannt wird, in der
Regel kurzlebig ist und unter Emission der Strahlung wieder zum Zustand A, der
‚nicht angeregt’ genannt wird, zurückkehrt. Es existieren Bewegungs-
(Translations-), Rotations-, Schwingungs,- und Elektronenzustände, die je
energiereicher die zu absorbierende Strahlung wird, nacheinander in dieser
Reihenfolge angeregt werden, d.h. Strahlung, die Elektronen anregt, ist
energiereich genug, um gleichzeitig auch Schwingungen, Rotationen und
Translationen auszulösen. Auch andere Energieformen als Strahlung, thermische
oder elektrische, können eine Anregung bewirken.
|
|
|
Abb.11 Elektromagnetisches Spektrum |
Isaac Newton (1643-1727) wusste noch nichts von
Energiezuständen der Materie, als er 1672 das weiße Sonnenlicht mit einem
Prisma in das Spektrum des sichtbaren Lichtes (Abb.11)
brach, ebensowenig Joseph
von Fraunhofer (1787-1826), als er 1814 mit dem ersten Spektroskop der Welt in
diesem Spektrum schwarze Linien entdeckte, und auch die Begründer der
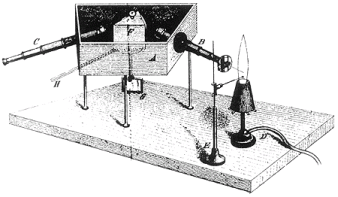
Spektralanalyse Robert Bunsen (1811-1889) und Robert Kirchhoff (1824-1887), die
1868 den ersten Spektralapparat der Welt (Abb.12) konstruierten, erlebten diese
Erkenntnisse des 20.Jahrhunderts nicht mehr.
|
|
|
Abb.12 Spektralanalysenapparat |
Bunsen und Kirchhoff verdampften Elemente in einer heißen
Flamme, fokussierten das von den heißen Dämpfen emittierte Licht auf ein
verstellbares Prisma (F) und betrachteten die projezierten Emissionsspektren mit
dem Spektroskop (C) ihres Apparates. Sie sahen sogenannte Linienspektren
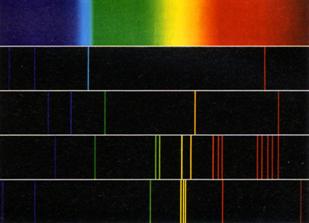
(Abb.13) mit wohldefinierten farbigen Linien und breiten dunklen Zwischenräumen,
kein kontinuierliches Farbband (Kontinuumspektrum) wie das Sonnenspektrum, und
entdeckten, dass alle Spektren verschieden und jedes für sein Element
charakteristisch war. Abb.13 zeigt unter dem Kontinuumspektrum der Sonne die
Linienspektren von Wasserstoff (H), Helium (He), Barium (Ba) und Quecksilber
(Hg). Jede Linie gehört zu einer definierten Wellenlänge, was indes noch nicht
der Sprachgebrauch um 1860 war.
|
|
|
Abb.13 Kontinuumspektrum und Linienspektren |
Weder Abb.11 noch
Abb.13 zeigen das Sonnenspektrum so,
wie es Fraunhofer mit hoher Auflösung in seinem Spektroskop sah. Durch
zahlreiche schwarze Linien unterbrochen gleicht es dem Negativ eines
Linienspektrums. Bunsen und Kirchhoff machten die faszinierende Entdeckung, dass
zahlreiche der schwarzen Linien mit den farbigen in ihren Elementspektren
deckungsgleich waren, und lieferten schließlich auch die Erklärung.
Es war die Geburtsstunde der Absorptionsspektroskopie.
Sie postulierten: Das Kontinuumspektrum der Sonne
resultiere aus den sich überlagernden Linienspektren einer Vielzahl thermisch
angeregter Elemente. In der kälteren Korona gebe es Gaswolken einiger dieser
Elemente, die noch nicht zur Strahlung angeregt seien, die aber aus dem
Sonnenlicht die Energie zur eigenen Anregung absorbieren könnten. Die
Absorption von definierten Energieportionen aus der Gesamtstrahlung manifestiere
sich in den schwarzen Unterbrechungen des Kontinuumspektrums, die mithin
Absorptionslinien seien. Jede Materie absorbiere aber wohl diejenige
Strahlungsart, die sie auch emittieren könne, dies erkläre die
Deckungsgleichheit der Linien.
Dieses Kirchhoffsche Strahlungsgesetz lautet moderner
ausgedrückt: Emittiert angeregte Materie oder spezieller ein angeregtes Element
Strahlung einer bestimmten Wellenlänge, so kann dieselbe Materie bzw. dasselbe
Element auch Strahlung derselben Wellenlänge absorbieren.
Die Spektroskopie diente in der Folge als qualitative
Methode emissions- wie absorptionstechnisch eingesetzt zur Entdeckung
zahlreicher Elemente und leistete ihren Beitrag bei der Aufstellung des
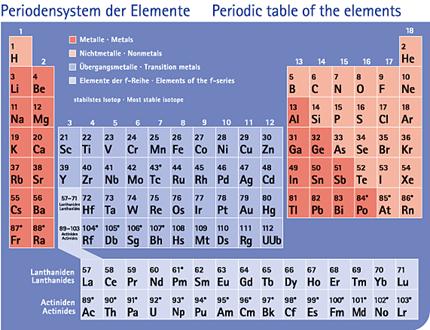
Periodensystems (Abb.6).
Die erste quantitative Anwendung war die Kolorimetrie
(Farbmessung), ein absorptionsspektroskopisches Verfahren, bei dem als
Messinstrument weiterhin das Auge diente. Nicht nur Elemente in der Gasphase,
sondern auch chemische Verbindungen in Lösung absorbieren Licht und in beiden Fällen
ist das Ausmaß der Absorption auf einer bestimmtem Wellenlänge proportional
abhängig von der Konzentration der absorbierenden Spezies und von der Länge
des Weges, den das Licht durch das Gas oder die Lösung zurücklegen muss. Diese
Abhängigkeiten sind Inhalt des Lambert-Beerschen Gesetzes, zu dem Johann
Heinrich Lambert (1728-1777), der schon 1760 die Abhängigkeit von der
Durchstrahlungsdicke entdeckte, und August Beer (1825-1863), der 1854 die
Konzentrationsabhängigkeit fand, beitrugen. In der Kolorimetrie vergleicht man
bei identischen Durchstrahlungsdicken eine Lösung unbekannter
Spezieskonzentration solange mit Lösungen bekannter Spezieskonzentrationen, bis
sich eine Farbgleichheit ergibt, aus der sich nach dem Lambert-Beerschen-Gesetz
die gesuchte Konzentration ableitet.
Das Kolorimeter, richtiger hätte es allerdings dem
Messinstrument Auge geschuldet Koloriskop (skopein (gr.): betrachten) heißen müssen,
ist ein Vorläufer des Photometers aus der Familie der Absorptionsspektrometer.
Ein Spektrometer ist im Gegensatz zu einem Spektroskop mit einer technischen
Licht- bzw. Strahlungsmessung ausgestattet, z.B. einer Photozelle, die Strahlung
in messbaren elektrischen Strom umwandelt, wodurch es Intensitäten, auch sehr
kleine, exakt zu detektieren vermag. Die andere wesentliche Innovation liegt in
der Verwendung monochromatischer Strahlung einer einzigen Wellenlänge, welche
merklich genaueres Messen als polychromatische Strahlung aus einem ganzen
Spektrum gestattet. Das Photometer ebnete den Weg zur Bestimmung von
Analytkonzentrationen im Spurenbereich und bot mit einem geeigneten Detektor
auch den Einstieg in die Absorptionspektrometrie mit anderen elektromagnetischen
Strahlungen als sichtbarem Licht.
Kolorimeter waren seit der Jahrhundertwende verfügbar,
wurden ab 1920 technisch stetig verbessert und deshalb bisweilen auch Photometer
genannt. Echte Photometer wurden aber erst ab 1945 kommerziell angeboten,
technische Hürden beim Bau von leistungsstarken Präzisionsmonochromatoren und
der 2.Weltkrieg verhinderten einen früheren Start.
Bereits Anfang der 30er-Jahren fand hingegen die
klassische qualitative Spektralanalyse ihre kommerzielle Fortsetzung.
Quantitativ arbeitende Flammenemmissionsspektrometer, auch Flammenphotometer
genannt, eroberten die Labore. Auch hier war das Messinstrument Auge des
Spektroskops von der Photozelle abgelöst worden. Auch hier filterte ein
Monochromator einzelne Linien bzw. Wellenlängen heraus, der es allerdings
bedeutend leichter hatte als sein Pendant im Photometer. Da sich die
Flammenphotometrie auf die quantitative Bestimmung weniger in einer chemischen
Flamme anregbarer Elemente beschränkte, hatte der Monochromator nur wenige sich
überlagernde Linienspektren auseinander zu dividieren. Es waren vor allem die
thermisch leicht anregbaren Alkali- und Erdalkalimetalle, die sich nun in sehr
niedrigen Konzentrationen bestimmen ließen. Die Stärke des Messsignals hängt
in der quantitativen Emissionsspektrometrie proportional von der Zahl der
angeregten strahlenden Elementteilchen ab, welche wiederum proportional mit der
Konzentration des Elements in der zu verdampfenden Probe korreliert.
Die Messprinzipien der Photometrie und der
Flammenphotometrie verdeutlichen noch einmal die Abb.14 und
15, bevor wir um das
Basiswissen des optischen Messwesens reicher zur Laborgeschichte der
Elementanalytik in Hessens Landwirtschaftlichen Versuchsstationen zurückkehren.
Es sind keine Absolutverfahren, sondern sie müssen beide immer mit
entsprechenden Vergleichsproben bekannten Gehaltes kalibriert werden.
|
|
|
|
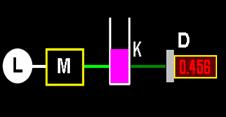
Abb.14 Photometrie |
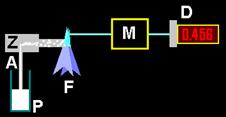
Abb.15 Flammenphotometrie |
(Legende
Abb.14: L = Lichtquelle, M = Monochromator, K = Küvette mit Probe, D = Detektor)
(Legende Abb.15: P = Probe, A = Ansaugung, Z = Zerstäuber, F = Flamme, M + D wie vor
)
![]()
Das zweite halbe Jahrhundert
Kaum anderes als steigende Probenzahlen war aus den
Versuchsstationen in Sachen Elementanalytik aus dem ersten Jahrzehnt des
20.Jahrhunderts zu vermelden gewesen. Das zweite schickte sich an, so
fortzufahren - es wurden neue, qualitativ z.T. bedenkliche Mischfuttermittel
produziert, die der dringenden Überwachung bedurften, neue Stickstoffdünger drängten
nach der gelungenen Ammoniaksynthese aus Stickstoff und Wasserstoff auf den
Markt - , als der 1.Weltkrieg die Forschungs- und Untersuchungstätigkeiten jäh
beschränkte. Der personelle Aderlass durch Einberufungen war hoch. In Kassel
reduzierte sich die Belegschaft auf weniger als ein Drittel, und die Situation
änderte sich auch nach Kriegsende nur unwesentlich. Von den Einberufenen kehrte
niemand zurück, und die inflationäre und durch Reparationen belastete
Wirtschaftslage verhinderte Neueinstellungen. Noch 1930 belief sich die Zahl der
wissenschaftlichen und technischen Mitarbeiter auf nur sieben. In Darmstadt war
die Ausgangslage besser, da es, von Paul Wagner forciert, zuletzt mehr
Mitarbeiter als in Kassel gegeben hatte, doch auch hier erfolgte eine Zäsur. In
den 20er-Jahren im nach Kriegsende aus dem Großherzogtum entstandenen
Volksstaat Hessen erholte sich die Station personell etwas schneller, sie
profitierte nach der Währungsreform 1923 von der besseren wirtschaftlichen Lage
im süddeutschen Raum und der Nähe zu den wesentlichen Produzenten
chemisch-landwirtschaftlicher Produktionsmittel.
Der Krieg brachte aber auch andere als personelle Nöte.
So konnte die chemische Industrie eine Zeitlang nicht ausreichend Citronensäure
produzieren, was die Phosphorbestimmung in Düngern einschränkte. Neue
Untersuchungsmethoden waren bei Futtermitteln gefragt. Immer öfter war die
Untersuchung von Futterkalken und ab Mitte des Krieges besonders von Tierkörpermehlen
gefragt, traurig machender ‚Nachschub’ von der Westfront, durch den sich
neben der deutlichen Zunahme von Haferuntersuchungen der Krieg am Labortisch
manifestierte. Neben Stickstoff wurde in Futtermitteln deshalb jetzt häufiger
auch Calcium (Ca) bestimmt.
Üblich war die Fällung als Oxalat oder Pikrolonat mit
gravimetrischer oder titrimetrischer Endbestimmung. Die Fällung mit Pikrolonsäure
eignete sich auch als mikrogravimetrische Methode, nachweisbar waren noch 10-20
mg Ca pro Liter, oder nach Auflösung des Pikrolonats in Salpetersäure und
Erzeugen einer blutroten Lösung bei Zugabe von Natronlauge für die
Kolorimetrie mit einer der Mikrogravimetrie vergleichbaren Nachweisstärke.
Mit Oxalsäure ((CO2H)2) und dem
Pyrazolinderivat Pikrolonsäure ((C3N2HO)(C6H4NO2)(CH3)
(NO2)) begegnen wir erstmals organischen Reagenzien in der
Elementanalytik. Da es zumeist Organika sind, die die Elemente per
elementorganischer Verbindungen zur Absorption von Licht oder UV-Licht und zur
kolorimetrischen oder photometrischen Bestimmbarkeit befähigen, sei hier
besonders auf sie verwiesen. Das intensivere Suchen nach geeigneten organischen
Substanzen zu Verbindungsbildung oder Komplexierung nahm zu dieser Zeit seinen
Anfang und bescherte uns bis heute eine reiche Palette. Ihr wichtigstes Attribut
ist ihre Spezifität für Elementpartner wie Bestimmungsverfahren. Von Reiz an
der Suche war, dass chemischer Sachverstand allein nicht zum Ziele führen
musste, sondern vielfach Empirie half.
Die von Not und gesellschaftlichen Veränderungsprozessen,
aber auch Innovation und Ideenreichtum geprägte Weimarer Zeit spiegelte sich
im Alltag der Versuchsstationen wieder. Die Not zeigte sich in den
Untersuchungszahlen. 1923 wurden in Kassel z.B. nur 17% der im Einzugsbereich
eingesetzten Düngemittel und nur 6% der Futtermittel untersucht. Die Landwirte
hatten nicht das Geld, die Untersuchungen zu bezahlen.
Dabei wäre eine höhere Zahl von Kontrolluntersuchungen
insbesondere bei Mischfuttermitteln dringend geraten gewesen, um vermehrt
auftauchende unseriöse Produkte vom Markt entfernen zu helfen. Seit 1920 gab es
zwar eine Mischfuttermittelverordnung, die aber, vom Charakter her lediglich
eine Sollvorschrift, wenig befolgt und vom ‚Verband der Landwirtschaftlichen
Versuchsstationen im Deutschen Reiche’ rigoros abgelehnt wurde. Emil Haselhoff
(1862-1948), von 1902 bis 1930 Direktor in Kassel und einer der wichtigen
Tierernährungswissenschaftler der 20er-Jahre, sagte seine Meinung unverblümt:
„Das beste wäre ja gewesen, diese Verordnung hätte nur einen einzigen
Paragraphen gehabt, der gelautet hätte: Die Herstellung von Mischfutter ist
verboten.“ 1927 gab es dann das erste deutsche Futtermittelgesetz, dass von
allen Seiten mehr Akzeptanz fand und letztlich auch den Freiraum für die
Entwicklung weiterer Futtermittelsorten schuf.
Für die Elementanalytik gab es Arbeit, als die
Futtermittelhersteller neue Mineralstoffmischungen erprobten. Sie versuchten
dabei neue wissenschaftliche Erkenntnisse über einen noch nicht ganz
verstandenen Mineralstoffwechsel umzusetzen. Die Mischungen enthielten Phosphor
(P), Calcium (Ca), Magnesium (Mg,) Kalium (K), Natrium (Na), Eisen (Fe), Zink
(Zn) und Kupfer (Cu). Gefragt war bei den Untersuchungen kein extraktiver,
sondern der Gesamtgehalt.
Die Überprüfung sicherte nur die Deklaration ab, sie
bot keine Beratung hinsichtlich geeigneter Mischungen. Dazu war das Wissen über
richtig abgestimmte Dosen an Mineralnährstoffen und Spurenelementen noch zu
unausgereift. Es wundert deshalb nicht, dass sich die Geister an diesen
Mischungen erneut schieden, weil nachprüfbare Erfolge selten waren, im
Gegenteil sogar negative Gesundheitsfolgen bekannt wurden, was aus heutigem
Wissen über Antagonisten unter den Nährelementen nicht erstaunt, bei deren
Unausgewogenheit Mangelerscheinungen die Folge sein können. Die
Mineralstoffmischungen dieser Zeit verschwanden deshalb oft genau so schnell
wieder vom Markt wie sie auf ihm erschienen waren.
Emil Haselhoff, der über 600 Veröffentlichungen
publizierte, verfasste in den Jahren 1924-29 gemeinsam mit dem Göttinger
Chemiker Edwin Blanck (1877-1953) auch ein vierbändiges ‚Lehrbuch der
Agrikulturchemie’, das ein Standardwerk seiner Zeit wurde. Betrachten wir in
Anlehnung daran, wie z.B. Eisen kolorimetrisch bestimmt wurde.
Gefärbte Verbindungen, die sich für die Kolorimetrie
eigneten, waren von dreiwertigem Eisen z.B. mit Ammoniumthiocyanat (NH4SCN)
oder Sulfosalicylsäure (C6H3)(OH)(CO2H)(SO3H))
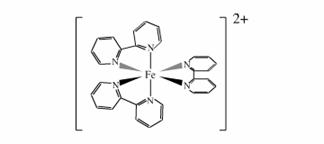
und von zweiwertigem Eisen mit Dipyridyl ((C5H5N)2)
(Abb.16) bekannt. Zu einer Gesamteisenbestimmung musste vorab entweder das
zweiwertige oxidiert oder das dreiwertige Eisen reduziert werden.
|
|
|
Abb.16 Komplexes Eisen-II-tris(α,α’-dipyridyl)-ion |
Vertiefen wir etwas die letztgenannte Bestimmung. Die
Mineralstoffmischung wurde zunächst einem sauren Aufschluss in konzentrierter
Salzsäure unterzogen oder im Muffelofen verascht und die Asche in
konzentrierter Salzsäure aufgenommen. Ob der nasse oder der trockene Aufschluss
geeigneter wäre, wurde kontrovers diskutiert. Heute würde bei einer
Mineralstoffmischung wohl eher der nasse Aufschluss gewählt, damals standen
nicht so reine Chemikalien zur Verfügung, und so hatte der
kontaminationsfreiere trockene Aufschluss seine Fürsprecher. Die salzsaure Lösung
wurde eingedampft und der Rückständ in verdünnter Salzsäure aufgenommen. Bei
Vorhandensein unlöslicher Anteile sah das Verfahren auch noch eine Filtration
vor. Mit Zugabe von etwas Wasserstoffperoxid (H2O2) ließ
sich noch vorhandenes zweiwertiges Eisen in dreiwertiges überführen, das dann
mit verdünnter Kalilauge als Eisenhydroxid (Fe(OH)3) gefällt wurde.
Enthielten die Mischungen Aluminium (Al) und/oder Mangan
(Mn) war eine vorherige Abtrennung unerläßlich. Das abfiltrierte Hydroxid
wurde in verdünnter Salzsäure gelöst, die Lösung zur Reduktion des Eisen in
die zweiwertige Form mit Natriumsulfit (Na2SO3) versetzt,
das Farbreagenz Dipyridyl zugegeben und die Lösung auf ein definiertes Volumen
aufgefüllt. Mit Vergleichslösungen bekannter Eisengehalte, der zu vermessenden
Lösung in Säure- und Reagenziengehalt angepasst, konnte nun das Messverfahren
(Abb.17) beginnen.
Die Kolorimetrie reifte in Kassel und Darmstadt in den
20er-Jahren zum Routineverfahren. Neben dem Einsatz bei der Kontrolle von
Mineralstoffmischungen, Düngemitteln, Wässern und Lebensmitteln wurden
wissenschaftliche Arbeiten begleitet. Systematische Analysen von
Futtermittelaschen sollten die Kenntnisse über grundsätzliche Mineralstoffvorräte
verschiedener Futtermitteltypen verbessern oder Pflanzenanalysen aus z.B. Bor-
(B), Chrom- (Cr), Kupfer- (Cu), Mangan- (Mn) und Schwefeldüngungsversuchen (S),
teilweise vor dem 1.Weltkrieg konzipiert und begonnen, das Wirkvermögen dieser
Elemente klären. Initiert wurden solche Versuche oft durch unerklärliche
Pflanzenkrankheiten, indes verliefen sie so ergebnislos, dass Haselhoff 1928
noch erklärte: „Es besteht keine Veranlassung, den ‚Reizstoffen’ Mangan
und Kupfer besondere Bedeutung für die Ertragssteigerung im praktischen Betrieb
zuzuschreiben.“ Spätere Forschergenerationen sollten eben auch noch etwas zu
entdecken haben.
Nicht immer ist aus dem vorhandenen historischen
Schriftwerk ersichtlich, ob für die Elementanalytik der 20er-Jahre jeweils eine
kolorimetrische Methode zur Hand war, wenn aber, dann ist ziemlich sicher, dass
sie anderen Methoden wegen ihrer Selektivität, Reproduzierbarkeit und
ihres Probendurchsatzes vorgezogen wurde. Elektrochemische Methoden faradayscher
Prägung, denen Redoxprozesse an Elektroden zugrundeliegen (Elektrogravimetrie,
Coulometrie, Voltammetrie, Amperometrie), wie nichtfaradayscher Prägung
(Konduktometrie, Potentiometrie) ergänzten die apparative anorganische
Analytik, erreichten aber in der Routine nie die Bedeutung des optischen
Verfahrens. Unerwähnt sollen auch nicht die in den 20er-Jahren vermehrt
entwickelten nephelometrischen Bestimmungen bleiben, die auf der
konzentrationsabhängigen Beugung von Licht durch gezielt erzeugte kolloidale Trübungen
von Analytlösungen beruhen. Sie waren zwar ebenso empfindlich, schnell und
selektiv wie kolorimetrische Verfahren, ließen sich aber wegen allerlei Störeinflüsse
nur schlecht reproduzieren und konnten sich nicht durchsetzen.
Streichen wir die Bedeutung der Kolorimetrie für das
landwirtschaftliche Untersuchungswesen dieser Zeit noch mit einem weiteren
konkreten Beispiel heraus, der auch für die spätere Photometrie wichtig
bleibenden Bestimmung des Phosphors mit der Molybdänblaumethode. Die Bestimmung
ist für nahezu alle Probenarten einer Landwirtschaftlichen Versuchsanstalt
einsetzbar und für Aufschlusslösungen ebenso geeignet wie für verschiedene
Extrakte.
Molybdän (Mo) tritt in seinen Verbindungen in den
Oxidationsstufen II bis VII auf und bildet verschiedene Mischoxide, in denen es
in mehr als einer Oxidationsstufe vorkommt. Eines dieser Mischoxide ist das
Molybdänblau ((MoO2)(MoO3)4), das seine
intensive blaue Farbe bei Verdünnung mit Wasser verliert. In Gegenwart von
Phosphat bleibt die Blaufärbung jedoch durch Verbindungsbildung stabil und ist
zwischen 0,01 und 10 mg/l proportional zur Konzentration des Phosphats. Das
Reagenz lässt sich in schwefelsaurer Lösung durch Reduktion von MoO3
mit elementarem Molybdän erzeugen. Eine Schwierigkeit ist die völlig
gleichartige und ebenso proportionale Reaktion mit Arsenat. Waren Phosphor und
Arsen (As) in vergleichbarer Menge in einer Probe enthalten, empfahlen die
Autoren die kolorimetrische Summenbestimmung beider Parameter, die nachfolgende
Einzelbestimmung des Arsens z.B. mittels einer jodometrischen Titration und
letztlich die Phosphorbestimmung aus der Differenzbildung.
|
|
|
Abb.17 Kolorimeter um 1920 |
In den Kasseler Jahresberichten tauchen zum Ende des
Jahrzehnts zur Freude des Chronisten auch wieder einige besondere Analyten mit
genaueren Gehaltsangaben auf. Von 37 Arsenbestimmungen an Superphosphatdüngern
mit Gehalten zwischen 91 und 335 mg/kg wird 1929 berichtet. Das Arsen ist
Kontaminant aus der zum technischen Aufschluss der Phosphate eingesetzten
Schwefelsäure. Weiter ist zu lesen, dass diese Konzentrationen keine Wirkungen
auf das Pflanzenwachstum erkennen ließen. 1931 werden wieder einmal
Wasseruntersuchungen erwähnt. Neben einigen Eisenanalysen werden in einer Probe
auch 0,44 mg/l Mangan (Mn) bestimmt. In einer Lebertranprobe wird ein erhöhter
Zinkgehalt (Zn) nachgewiesen und in einer Weizenmehlprobe Quecksilber (Hg),
leider ohne Gehaltsangabe oder Bemerkung, ob von erhöhtem Gehalt, aber mit dem
Hinweis, dass das Schwermetall aus Saatgutbeizmitteln stammen müsse.
Aus der Landwirtschaftlichen Versuchsstation in Kassel war im übrigen 1922 eine Landwirtschaftliche
Versuchsanstalt geworden. Nicht so
genau datierbar, aber noch in den 20er-Jahren erfolgte die gleiche Namensänderung
in Darmstadt.
Die elementanalytischen Untersuchungstätigkeiten beider
Anstalten wurden um 1930 nach wie vor von den Düngemittelanalysen auf
Stickstoff, Phosphor und Kalium bei gestiegenem Kaliumanteil und den
Futtermittelanalysen auf Stickstoff dominiert, aber neben den ersten
Gehversuchen zu anderen Mineralstoffen erwachte das Interesse an der Nährstoffsituation
der Böden.
Hans Wießmann, der 1930 Emil Haselhoff als Leiter in
Kassel ablöste, hielt auf dem Verbandskongress 1934 ein Grundsatzreferat über
Düngung, in dem er darauf abhob, dass die Nahrungsproduktion immer noch nicht
das Vorkriegsniveau erreicht habe, obwohl die doppelte Menge an Stickstoff und
erheblich mehr Kalium gedüngt würden. Seine Schlussfolgerung ging zwar
letztlich in Richtung einer gestörten Bodenfruchtbarkeit durch
Humusunterversorgung, mit der Ertragsssituation war aber auch die Frage nach
einer generell ausreichenden und im Mengenverhältnis adäquaten
Mineralstoffversorgung verknüpft.
Der politische Umschwung 1933 und in der Folge die
politische Forderung nach einer weitgehend autarken und qualitativ hochstehenden
Versorgung mit landwirtschaftlichen Gütern versetzte die Versuchsanstalten in
ganz Deutschland dann in kurzer Zeit personell und materiell in die Lage, ein
umfangreiches systematisches Bodenuntersuchungswesen aufzunehmen. So gab es in
Kassel 1937 wieder 25 MitarbeiterInnen im Labor und im Landwirtschaftlichen
Versuchswesen, darunter 8 wissenschaftliche, und in Darmstadt sogar 47, darunter
6 wissenschaftliche.
|
|
|
Abb.18 Agrikulturchemisches Labor in Kassel 1935 |
Zwischen 1936 und 1944 wurden in Deutschland
elementanalytisch 5,7 Millionen Phosphor- und 500.000 Kaliumgehaltsbestimmungen
durchgeführt, daneben mit anderen als elementanalytischen Verfahren
(Keimpflanzenversuch und pH-Bestimmung) weitere 500.000 Kalium- und 7,2
Millionen Kalkbedarfsbestimmungen, hier aber miterwähnenswert, um die Dimension
des Großprojektes zu veranschaulichen. 4,9 Mio. Phosphoruntersuchungen
entfielen auf die Kriegsjahre 1941-44, in denen der sparsame Umgang mit den
Phosphatreserven oberste Düngepflicht war, da der Import von Rohphosphat
zunehmend zum Erliegen kam. Jede Versuchsanstalt musste für die als
‚Bodenuntersuchungs-Sonderaktion’ ausgelobte Maßnahme statt einiger hundert
Böden pro Jahr wie bis Mitte der 30er-Jahre einige tausend bearbeiten (Beispiel
Kassel: 1936: 364; 1937: 711; 1938: 2688; 1939: 9177; 1940: 15937) und wäre
ohne physikalische Messmethoden an dieser Aufgabe gescheitert. Die
Bodenuntersuchung, wie wir sie heute als eine der zentralen Aufgaben unserer
Versuchsanstalten kennen, hatte sich etabliert.
Unter Ludwig Schmitt, der die Leitung der Versuchsanstalt
1933 übernahm und später langjähriger Präsident des Verbandes wurde, konnte
1936 in Darmstadt das erste Flammenphotometer (Abb.15) erworben werden, und die
Emmissionsspektrometrie bestand die Feuertaufe bei den Kaliummassenbestimmungen
erfolgreich. Sie ist für diesen Anwendungszweck auch heute noch die Methode der
Wahl. In Darmstadt wurden in den Kriegsjahren 14.000 Böden auf laktatlösliches
Kalium untersucht, in Kassel, wo noch kein Flammenphotometer zur Verfügung
stand und kolorimetrisch gearbeitet wurde, kamen 3000 Böden zur Kaliumanalyse
auf den Labortisch.
Die in den 30er-Jahren in der Verbandsarbeit entwickelte
kolorimetrische Methode nutzt die quantitative Fällbarkeit des Kaliums mit
einer Mischung aus Kobaltnitrat (Co(NO3)2) und
Natriumnitrit (NaNO2) unter oxidativen Bedingungen zum komplexen
Kaliumnatriumkobaltnitrit (K3Co(NO2)6 x Na3Co(NO2)6).
Das gefällte Salz ist in Natronlauge löslich, bildet mit Sulfanilsäure und
Naphtylamin einen Farbkomplex und ist so kolorimetrisch analysierbar. Das
Methodenbuch des Verbandes, in dem diese Vorgehensweise beschrieben wird, ist
das erste, das sich mit Bodenuntersuchungen beschäftigt. Beachtenswert ist,
dass für Bestimmungen verschiedener Schwermetalle hier bereits das
Aufschlussverfahren mit Königswasser (3:1-Mischung aus Salzsäure und Salpetersäure)
vorgeschlagen wird, das noch heute im Landwirtschaftlichen Untersuchungswesen in
der chemischen Aufarbereitung mineralisch dominierter Probenspezies seinen
unangefochtenen Stammplatz hat.
Das Erstaunen des Chronisten gegenüber dem
elementanalytischen Wissen der Altvorderen kam schon einmal zur Sprache, als es
Spurenanalytik im Jahre 1900 zu entdecken gab. Wenn eine Tatsache nach dem
Studium der alten Dokumente noch einmal einer besonderen Betonung bedarf, dann
die, dass es wahrscheinlich alles früher gab, als es der Chronist aufspüren
konnte.
Neben soviel Bodenuntersuchung und Massenanalytik in den
30er- und 40-er-Jahren brachten die hessischen Elementanalytiker aber auch in
die Angewandte Forschung weiterhin ihre Arbeit ein. So konzipierte die Anstalt
Darmstadt 1937 die wohl bis dato ausgedehntesten Düngungsversuche weltweit zum
Zwecke der umfassenden und kurzfristigen Prüfung der in dieser Zeit als
Wundermittel der Düngung gepriesenen Gesteinsmehle, die Anbieter langfristig
verfügbarer mineralischer Makro- und Mikronährstoffe sein sollten.
|
Element |
Gehalte |
Element |
Gehalte |
Element |
Gehalte |
|
Si |
14 - 31% |
Ca |
0,1 - 18% |
Mg |
0,7 - 10% |
|
Fe |
0,7 - 8,4% |
K |
0,7 - 3,7% |
N |
0,1 - 0,2% |
|
P |
u.B. - 0,9 % |
Al |
u.B. - 0,1% |
Na |
u.B. - 0,1% |
|
Sr |
u.B. - 0,1% |
Mn |
u.B. - 0,1% |
Ni |
u.B. - 0,1% |
|
Ti |
u.B. - 0,1% |
Ba |
u.B.- 0,01% |
Co |
u.B.- 0,01% |
|
Cr |
u.B.- 0,01% |
Cu |
u.B.- 0,01% |
V |
u.B.- 0,01% |
|
Ag |
u.B.- 0,001% |
B |
u.B.- 0,001% |
Mo |
u.B.- 0,001% |
|
Pb |
u.B.- 0,001% |
Sn |
u.B.- 0,001% |
Zn |
u.B.- 0,001% |
Abb.19 Elementgehalte in Steinmehlen aus Darmstädter Pflanzenbauversuchen
(u.B.
= unterhalb der Bestimmbarkeitsgrenze)
In 9 Gesteinsmehlen aus pflanzenbaulichen Versuchen
wurden auf Grund dieser Behauptungen nicht nur die Gehalte an Stickstoff (N),
Phosphor (P), Kalium (K), Calcium (Ca), Magnesium (Mg), Eisen (Fe) und Silicium
(Si) gravimetrisch, titrimetrisch oder kolorimetrisch bestimmt, sondern auch die
an Natrium (Na), Strontium (Sr), Aluminium (Al), Barium (Ba), Bor (B), Silber
(Ag), Kobalt (Co), Chrom (Cr), Kupfer (Cu), Mangan (Mn), Molybdän (Mo), Nickel
(Ni), Blei (Pb), Titan (Ti), Vanadium (V), Zinn (Sn) und Zink (Zn)
kolorimetrisch oder flammenphotometrisch erfasst. Die Vielfalt der beherrschten
optischen Bestimmungen sei damit eindrucksvoll vorgeführt.
Abb.19 zeigt
tabellarisch die ermittelten Gehalte als Bereiche, wobei ein 98%-reines
Quarzmehl bei der Aufstellung unberücksichtigt blieb.
Die Düngungsversuche mit 8-jähriger Laufzeit belegten
schließlich die ziemliche Wirkungslosigkeit der Gesteinsmehle im Vergleich zur
klassischen NPK-Mineraldüngung.
Die Effektivität von Düngung und Fütterung zu
steigern, avancierte in den ernährungskritischen Nachkriegsjahren und bis weit
in die 50er-Jahre hinein wieder zur Aufgabe mit der höchsten Priorität für
die Versuchsanstalten.
So wurde die Phase der umfassenden Bodenanalysen trotz
schwerer Gebäudezerstörungen an beiden Standorten auch nach Kriegsende
forciert fortgesetzt. Bis 1948 wurden z.B. in Darmstadt weitere rund 44.000, in
Kassel und Marburg, wohin die kurhessische Anstalt für ein Jahr ausgelagert
wurde, weitere rund 20.000 Böden auf ihren Kaliumgehalt untersucht.
Zunehmend deutlicher wurde der Agrarforschung in diesen
Jahren nun auch, dass die alleinige Ausbringung bzw. Verfütterung von Makronährstoffen
zur optimalen Ernährung von Pflanzen und Tieren nicht ausreichte. Die ernährungsphysiologische
Notwendigkeit anorganischer und organischer Mikronährstoffe wurde mehrheitlich
anerkannt. So unterbreitete auch Ludwig Schmitt 1954 als Präsident des
,Verbandes der Deutschen Landwirtschaftlichen Untersuchungs- und
Forschungsanstalten’ dem damaligen Bundeslandwirtschaftsminister Lübke eine
Denkschrift über die vordringlichen Aufgaben der Versuchsanstalten, in der
ausdrücklich die ‚Ausweitung der Forschung über die Mikronährstoffversorgung’
im Rahmen der Förderung der Bodenfruchtbarkeit benannt wurde. In den
Fachgruppen des Verbandes entstanden nun die Methoden, mit denen sich die
anorganischen Analytiker der Anstalten den Spurenelementen in Böden, Düngemitteln,
Futtermitteln und Pflanzen widmen konnten.
Bor (B), Kupfer (Cu), Mangan (Mn), Molybdän (Mo) und
Zink (Zn) waren um 1950 als essentiell wichtig für die Pflanzenernährung
erkannt, bei Chlor (Cl) , Eisen (Fe), Natrium (Na) und Silicium (Si) bestanden
Hinweise.
Die nun fundierteren Kenntnisse der
Futtermittelhersteller zum Bedarf an Spurenelementen in der Tierernährung
beendeten auch den jahrzehntelangen Widerstand der Versuchsanstalten gegen die
Mineralfuttermischungen des Handels und führten zu Kontrollverträgen. Bis 1955
verzehnfachten sich die Produktionsbetriebe und verdreifachte sich der Umsatz
der Branche, wovon die Anstalten profitierten. Die Futtermitteluntersuchung
widmete sich neben Stickstoff (N), Phosphor (P), Calcium (Ca), Kalium (K),
Magnesium (Mg), Schwefel (S) und Chlor (Cl) den erkannten essentiellen
Spurenelementen Eisen (Fe), Kobalt (Co), Kupfer (Cu), Mangan (Mn) und Zink (Zn).
Jod (J) kam bald, Selen (Se) um einiges später dazu.
In den 50er-Jahren wurden in Kassel und Darmstadt, beide
Anstalten nun im Bundesland Hessen angekommen, aber nach wie vor unabhängig
agierend, Photometer (Abb.20) eingeführt und viele kolorimetrische Verfahren
auf photometrische umgestellt.
Eine der damaligen spurenanalytischen Methoden, die
Bestimmung des Zinkgehaltes in Pflanzen, sei hier stellvertretend betrachtet. Es
begegnet uns dabei mit Dithizon (Diphenylthiocarbazon) (Abb.21) ein organisches
Reagenz, das als Farb- und Anreicherungsreagenz nun rund 3 Jahrzehnte das
spurenelementanalytische Labor prägen sollte und mancher Laborantin und manchem
Laboranten doch Stoßseufzer ob der zahlreichen Extraktionsschritte per
Scheidetrichter und der vom Schütteln derselben erlahmenden Armmuskeln
entlockte. Ganze Bücher wurden dem Reagenz gewidmet, das sowohl in sauren wässrigen
Lösungen als auch in organischen Lösungsmitteln stabile Verbindungen mit einer
Vielzahl von metallischen Elementen zu bilden vermag.
Die Pflanzenprobe wird zunächst bei maximal 550°C im
Muffelofen verascht, die Asche mit konzentrierter Salzsäure abgeraucht und der
Rückstand mit verdünnter Salzsäure zum Erhalt einer Analysenlösung
extrahiert. Dies ist auch heute noch eine gebräuchliche Aufschlusstechnik für
Futtermittel mit vorwiegend organischer Matrix. Wichtig ist, dass die
Veraschungstemperatur dem Analyt moderat angepasst ist, um Verluste von flüchtigen
Zinkhalogeniden zu vermeiden. Es gibt Analyten, wie Cadmium (Cd), bei denen
bereits 400°C die Obergrenze darstellen.
|
|
|
Abb.20 Photometer um 1960 |
Neben der landwirtschaftlichen Hochschule Hohenheim
leistete hier insbesondere die Darmstädter Versuchsanstalt unter Ludwig
Schmitt Anfang der 50er-Jahre essentielle Pionierarbeiten zu einer Reihe von
Elementbestimmungen. Gleichermaßen wurde an den Veraschungen wie den weiteren
Aufarbeitungsschritten gearbeitet.
Bei Zink gestalteten sich letztere wie folgt: Die schwach
alkalisch gemachte Analysenlösung wird in einem Scheidetrichter mit einer Lösung
von Dithizon in Tetrachlormethan extrahiert. Dabei gehen Zink und andere Metalle
als rot gefärbte Dithizonate in die organische Phase, die in einen zweiten
Scheidetrichter überführt wird. Die Extraktion ist mit frischen Portionen
Dithizonlösung solange zu wiederholen, bis die organische Phase sich nicht mehr
rot färbt. Die gesammelten Extrakte werden nun mehrfach mit verdünnter Salzsäure
ausgeschüttelt, wobei Zink und die anderen Metalle in die saure wässrige Phase
wechseln. In einem dritten Scheidetrichter werden die gesammelten salzsauren
Extrakte auf einen pH-Wert zwischen 4,5 und 5,0 eingestellt und mit
Natriumthiosulfatlösung versetzt, um dann erneut erschöpfend mit Portionen
Dithizonlösung extrahiert zu werden. Im Unterschied zur ersten Extraktion
wechselt unter diesen Bedingungen allein das Zink in die organische Phase. Die
Konzentration des Zinkdithizonates konnte im Vergleich mit Eichlösungen
bekannter Zinkkonzentration nun entsprechend dem Lambert-Beerschen Gesetz
photometrisch bei einer Absorptionswellenlänge von 538 nm bestimmt werden.
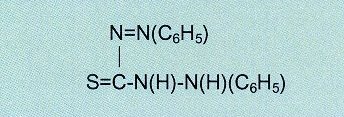
Abb.21 Dithizon
Für viele andere Metalle (z.B. Silber (Ag), Blei (Pb),
Cadmium (Cd), Kupfer (Cu), Nickel (Ni) und Quecksilber (Hg)) wurden in diesen
Tagen spezifische Trennverfahren auf der Basis der Anreicherung ihrer
metallorganischen Verbindungen zwischen nicht mischbaren Phasen entwickelt. Das
favorisierte organische Reagenz in Darmstadt und Kassel war das Dithizon, aber
es kamen für die Photometrie auch andere wie Diethyldithiocarbaminsäure (Abb.22), bevorzugt bei Kupfer, oder Diacetyldioxim
(Abb.23), bevorzugt bei
Nickel, zum Einsatz.
Es 'duftete' immer reichlich nach flüchtigen
Chlorkohlenwasserstoffen im Spurenelementlabor. Der Arbeitsschutz musste erst
noch entdeckt werden. Zwei Jahrzehnte später fiel die Wahl des Lösungsmittels
aus Gründen geringerer Giftigkeit eher auf Dichlor- statt Tetrachlormethan.
Viel hat das die Laborluft nicht entlastet, es wurde dann aber vermehrt der vernünftige
Weg zu den Abzügen angetreten, deren Verbreitung in den 50er-Jahren noch zu wünschen
übrig ließ.
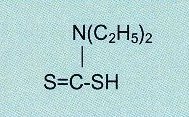
Abb.22
Diethyldithiocarbaminsäure
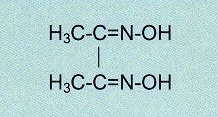
Abb.23
Diacetyldioxim
Neben der Isolierung eines Analyten eröffneten die neuen
Verfahren auch seine Aufkonzentrierung, indem die Volumina der extrahierenden
Phasen geringer gewählt wurden als die der zu extrahierenden. Zusammen mit der
technischen Verbesserung der Bestimmungsgrenzen durch die Photometrie waren nun
Gehalte um 0,01 mg/l und darunter auch in der Serienanalytik zugänglich. Es ist
deshalb wohl trotz früherer Vorstöße zu kleinsten Gehalten, die allesamt von
großem Aufwand und wenigen Proben gekennzeichnet waren, zulässig, in die
50er-Jahre den wahren Beginn der anorganischen Spurenanalyse zu legen.
Wenn von metallorganischen Reagenzien die Rede ist, muss
auch die Einführung der komplexometrischen Titration in Kassel und Darmstadt
gegen Ende des Jahrzehnts mit ihrer Bedeutung für die Düngemittel- und
Bodenanalytik vorgestellt werden. Die vom Schweizer Chemiker Gerold
Schwarzenbach (1904-1978) Mitte der 40er-Jahre entwickelte Methodik fand in den
Anstalten hauptsächlich Anwendung in der Calcium- und Magnesiumanalytik, deren
Schwierigkeit in der Trennung beider Elemente lag, und löste aufwendigere
gravimetrische und störanfälligere flammenphotometrische Verfahren ab. Auch
heute werden die Erdalkalielemente in Düngemitteln z.T. noch komplexometrisch
erfasst.
Der für diese Form der Titration zuerst aufgefundene und
bis heute bekannteste Komplexbildner ist das Dinatriumsalz der
Ethylendiamintetraessigsäure (EDTA) (Abb.24). Es vermag ein Metallkation über
sechs Kontaktstellen hoher Elektronendichte an zwei Stickstoff- und vier
Sauerstoffatomen koordinativ zu binden, was zu einem außerordentlich stabilen
Metallkomplex führt. Man spricht bei EDTA von einem mehrzähnigen Liganden.
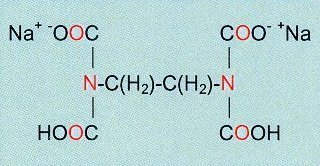
Abb.24
Dinatriumethylendiamintetraacetat (EDTA)
Zur komplexometrischen Titration ist neben einem starken
Komplexbildner noch ein spezifischer Farbstoff erforderlich, der sich ebenfalls
an das zu titrierende Element binden kann und in gebundener anders als in reiner
Form färbt. Schwarzenbach bot bereits eine Palette an Farbstoffen an, heute
sind über 50 geeignete Substanzen bekannt. Wird die
Element-Farbstoff-Verbindung mit einem starken Komplexbildner bekannter
Konzentration titriert, verdrängt er den Farbstoff aus der Metallbindung. Im
Moment der völligen Freisetzung des Farbstoffs ändert die Lösung die Farbe,
der Äquivalenzpunkt ist erreicht und die verbrauchte Menge Komplexbildner
liefert die Elementkonzentration.
Welche Entwicklungen im Nachkriegsjahrzehnt hatten noch
Auswirkungen auf die Elementanalytik?
In der Bodenuntersuchung, wo sich die jährlichen
Probenzahlen beider Anstalten weiterhin nach Zehntausenden rechneten, gesellte
sich zum Phosphor und Kalium das Magnesium. Bei den Mineraldüngern lichtete
sich der Markt. Weniger Produzenten bedeuteten allerdings keinen Rückgang der
Gesamtproduktion. Indes waren die Probenzahlen rückläufig, da die Produkte
qualitativ besser wurden, die Produzenten dies über Selbstkontrollverträge mit
dem VDLUFA absicherten und deshalb weniger Produktionskontrollen erforderlich
wurden. Hingegen expandierten die Mineralstoffuntersuchungen an
wirtschaftseigenen Düngemitteln und ersten Abfallstoffen der jungen
Industriegesellschaft, die in der Landwirtschaft eine Entsorgungsmöglichkeit
suchte. Dies führte gegen Ende des Jahrzehnts zu ersten Überlegungen, welche
Schadelemente diesen Frachten beiwohnen könnten. Der Anstieg der
Futtermitteluntersuchungen wegen der neuen Sortenvielfalt und der Ausweitung des
Handels wurde bereits angesprochen. Hinzu kam neu die Amtliche
Futtermittelkontrolle. Das Landwirtschaftliche Versuchswesen schließlich bemühte
sich, den Forschungsergebnissen zur ganzheitlichen Mineralstoffversorgung der
Pflanzen mit Makro- und Mikronährstoffen Rechnung zu tragen und war an beiden
Standorten besonders mit Bor-, Kupfer-, und Magnesiumdüngungsversuchen
Lieferant von Aufwuchsproben für die elementanalytischen Labore. Daneben
liefen natürlich auch die klassischen Versuche mit N, P und K weiter.
Von der allgemein zunehmenden Prosperität des Landes
profitierten auch die Anstalten. Nach dem Wiederaufbau und der Konsolidierung
begann mit dem ausgehenden Jahrzehnt eine Phase mit verstärkten Investitionsmöglichkeiten.
Personell und apparativ ging es aufwärts.
Beschließen wir das zweite halbe Jahrhundert
elementanalytischen Schaffens im Hessischen Landwirtschaftlichen
Untersuchungswesen mit den von Aufbruchstimmung und Optimismus geprägten
50er-Jahren, die auch der Chronist in Kindertagen recht faszinierend fand, und
beginnen wir das dritte mit den 60er-Jahren des vorigen Jahrhunderts und ihrem
Start in die Atomspektrometrie und Laborautomatisation.
Vorher geht es noch einmal hinein in die Physik der
modernen Analysentechniken, damit im letzten Kapitel das Lesen leichter fällt.
![]()
Von der Photometrie zur Atomspektrometrie
Wir waren im Kapitel über das ‚Licht im Dunkel der
Elementanalytik’ bis zur Emissionstechnik ‚Flammenphotometrie’ und bis
zur Absorptionstechnik ‚Photometrie’ vorgedrungen.
Abb.25 zeigt den in den 60er-Jahren in Kassel eingerichteten,
mit Probengebern und Dosierpumpen automatisierten Messplatz für die
Elementbestimmung in Bodenextrakten, der mit einem Flammenphotometer (links) für
die Kalium- und einem Photometer (rechts) für die Phosphorbestimmung bestückt
war. Das Photometer stammt wie das Photo allerdings aus den späten 70er-Jahren.
Während die Flammenphotometrie eine echte
atomspektrometrische Technik ist, das Messsignal entspringt einer atomaren
Eigenschaft, der Emission von Licht thermisch angeregter Atome, ist die
Elementbestimmung per Photometer ein Umweg über eine Eigenschaft einer
molekularen Verbindung eines Elements, der Absorption von Licht des sich dabei
anregenden Moleküls. Der Vorteil der Flammenphotometrie ist die schnelle,
direkte Verarbeitung von Analytlösungen, der gravierende Nachteil seine Beschränkung
auf die wenigen durch chemische Flammen anregbaren Elemente.
Lediglich Natrium, Kalium und die restlichen Alkalielemente
in Propan/- oder Butan/Luft-Flammen bei Flammentemperaturen um 800°C und
Calcium, Magnesium und die restlichen Erdalkalielemente in der
Acetylen/Luft-Flamme, erstmals eingesetzt in den 50er-Jahren, bei
Flammentemperaturen um 2300°C sind so messbar.
Der große Vorteil der Photometrie ist die Vielzahl der bis
zu sehr niedrigen Konzentrationen messbaren Elemente - geeignete Farbreagenzien
gibt es seit den 50er-Jahren in großer Zahl -, der erhebliche Nachteil liegt in
der langwierigen Aufarbeitung der Analytlösungen mit Anreicherung und Anfärbung
und häufiger noch Abtrennung anderer Elemente für eine störungsfreie Messung.
Die Vereinigung der Vorteile beider Methoden war natürliches Bestreben der
Forschung und datiert bis in die frühen 40er-Jahre zurück.
|
|
|
|
Abb.25 Messplatz mit Flammenphotometer undPhotometer |
|
Ziemlich bald schloss man ein Atomabsorptionsverfahren aus.
Es war zwar unproblematisch, Analyten in einer heißen Flamme zu atomisieren und
damit analog der Küvette mit Analytlösung (s. Abb.14) in einem Photometer eine
Zone zu schaffen, in der Licht aus einer Strahlungsquelle absorbiert werden
konnte, doch die Quantifizierung der Absorption scheiterte an dem Problem, dass
eine Flamme mit angeregter Materie genau auf der Absorptionswellenlänge
Strahlung emittiert (s. Kirchhoffsches Strahungsgesetz im vorletzten Kapitel). Folglich ist eine ungestörte Messung der vom Analyten
nicht absorbierten, durchgelassenen Strahlung auf der gewählten
Absorptionswellenlänge eigentlich unmöglich, da sich immer emittierte
Strahlung der gleichen Wellenlänge aufsattelt. Da man diese grundsätzliche
Schwierigkeit für unüberwindbar hielt, wurde zunächst jegliche Forschung zum
Messprinzip Atomabsorption eingestellt.
Im Fokus des Interesses stand nun, das Messprinzip
Atomemission auf schwerer anregbare Atome als Alkali und Erdalkalimetalle zu übertragen.
Sehr wohl bekannt war schon, dass hierzu die thermische Energie der heißesten
chemischen Flammen nicht ausreichen würde. Die Versuche konzentrierten sich auf
Verdampfungen im elektrischen Lichtbogen zwischen zwei Kohleelektroden, wobei
die Probe entweder als Feststoff, verrieben mit Kohle, oder über ein
Tauchverfahren flüssig in eine Vertiefung einer Elektrode eingebracht wurde.
Das klingt nach heutigen Maßstäben langwierig und war es auch, da die Kohlen
vor jeder neuen Messung präpariert werden mussten und auch die Dosierung hohe
Sorgfalt erforderte.
Was allerdings noch länger währte, war die
Probenvorbereitung. Da kein geeignetes Monochromator-Detektor-System zur Verfügung
stand, das in der Lage gewesen wäre, aus der Emissionslinienvielfalt aller
Probenbestandteile eine spezifische quantitative Messung einzelner Elemente zu
gestalten, schaltete man ein Procedere zur Abtrennung der Probenmatrix und
Anreicherung der Analyten vor, dass dem Prinzip der Probenvorbereitung zwischen
nicht mischbaren Phasen der oben geschilderten photometrischen Zinkbestimmung
mit Dithizon entsprach. In der letzten Phase wurde nur kein einzelnes Element
isoliert, sondern mehrere, eben die gewünschten. Nun wurde entweder eingedampft
für die Feststoffvariante oder die Phase direkt verwendet für die
Tauchvariante. Damit war die Vorbereitung jedoch keineswegs zu Ende, sondern das
Messprinzip, eine Intensitätsauswertung der Emissionslinien auf einer
Photoplatte erforderte, dass zumindest der Gehalt eines Elementes in der Phase
exakt bekannt sein musste. Zumeist entschied man sich für Eisen, dass nahezu in
jeder Probe in gut messbarer Menge anzutreffen war. Die Gehaltsermittlung
erfolgte in der Regel photometrisch über den oben beim kolorimetrischen
Verfahren schon geschilderten Dipyridylkomplex (s.
Abb.16).
Nun, mit einem so aufwendigen Analysengang hatte die
Atomemissionstechnik zu Beginn der 50er-Jahre keinerlei Vorteile gegenüber der
Photometrie und war ihr auch in der Nachweisstärke nicht überlegen. Es war
eigentlich nur bewiesen, dass mit einer entsprechend hohen thermischen Anregung
eine quantitative Messung per Atomemission je nach Analyt von rund 0,1 mg/kg bis
zu einigen 100 mg/kg möglich war. Nach Kenntnis des Chronisten wurde das
Kohlelichtbogenverfahren auch nie in einer landwirtschaftlichen
Untersuchungsanstalt eingesetzt.
Der große Durchbruch der Atomspektrometrie als routinemäßig
in der Element- und Spurenelementanalytik einsetzbares und rationelles Verfahren
gelang dann 1952 doch überraschenderweise auf dem Gebiet der Atomabsorption.
Laut Überlieferung bei der Gartenarbeit habe den englischen Physiker Alan Walsh
(1916-1998) die Idee der modulierten Strahlungsquelle ereilt. Entweder mit einem
mechanischen Zerhacker (Chopper) oder einer elektrisch aufgesattelten Frequenz lässt
sich der zu absorbierende Lichtstrahl dergestalt modulieren, dass ein auf diese
Modulation abgestimmter Detektor nur das Licht der Strahlungsquelle verwertet
und nicht mehr die bisher auf der Absorptionswellenlänge interferierende
Emission der Flamme bzw. der thermisch angeregten Probe.
Es dauerte nach Veröffentlichung des Prinzips 1955 noch ein
knappes Jahrzehnt, bis sich nach der Entwicklung stabiler Strahlungsquellen
(Hohlkathoden- und elektrodenlose Entladungslampen) und dem hilfreichen Ruf aus
der US-amerikanischen Medizin nach hohen Stückzahlen schneller, verlässlicher
Serumanalysen auf Calcium - photometrisch wie flammenphotometrisch nie ganz
zufriedenstellend gelöst - die neue Technik kommerziell lohnte und etablierte.
|
|
|
|
Abb.26 Flammen-AAS-Gerät um 1965 |
|
Die Atomabsorptionsspektrophotometrie, kurz AAS, trat ihren
Siegeszug an. Sie war und ist wie alle optischen Verfahren kein
Absolutverfahren, sondern bedarf der Kalibrierung mit Lösungen bekannter
Analytgehalte. Sie ist ein sequentielles Verfahren, ein Element wird nach dem
anderen gemessen. Lange war sie ein reines Flammenverfahren unter Einsatz der
schon bekannten Luft-/Acetylen-Flamme und anderer Brenngasgemische, von denen
vor allem die um 2800°C heiße Lachgas (N2O)-/Acetylen-Flamme weite
Verbreitung fand. Ohne Anreicherung lag die erreichbare
Bestimmungsgrenze der Flammen-AAS für einige gut bestimmbare Analyten wie
Cadmium um 0,02 mg/l in der Analysenlösung oder auch ‚ppm’ (‚parts per
million’), wie es nun lange Jahre anglophil hieß, bis schließlich das
heimatverbundene ‚mg/l’ oder ‚mg/kg’, wenn ein Feststoff die Bezugsgröße
ist, obsiegte.
So mancher Spurenanalytiker vermisst seine ‚ppm’,
verdeutlichte doch ein ‚1 Teil auf 1 Million’ dem zum Staunen ausersehenen
Laien so viel anschaulicher die schwer vorstellbare Dimension von 10 hoch minus
6. Und aus mg/kg konnte auch nicht das noch anschaulichere ‚1 Preuße pro München’
abgeleitet werden. Und bei ‚ppb' (‚parts per billion') war die Erklärung
‚1 Preuße pro Bayern' doch eine humorvolle Steigerungsform. Nun zugegeben, da
‚billion' ja doch ‚nur' eine 'Milliarde' ist, und es auch nicht so viele
Bayern gibt, liegt im deutschen µg/l bzw. µg/kg mehr Klarheit.
In diesen Mikrogrammbereich stieß die Flammen-AAS allerdings
nur vor, wenn im Labor ausreichend Dithizon oder vergleichbare Organika und
chlorierte Lösungsmittel vorrätig waren. Von der Doppelfunktion als
Anreicherungs- und photometrisches Farbreagenz blieb für Dithizon und
seinesgleichen nach Einführung der Flammen-AAS zumeist nur die erstere.
|
|
|
|
Abb.27 Flammen-AAS-Gerät um 1975 |
Abb.28 Brenner eines |
Doch schon nahte mit der flammenlosen Graphitrohr-AAS das
Ende der dithizongeprägten Aera. Entwickelt Mitte der 60er-Jahre und zur
kommerziellen Reife in den 70er-Jahren gelangt, eröffnete sie dem
Spurenanalytiker für einen Großteil des Periodensystems die Dimension ‚10
hoch minus 9’ für die durchsatzstarke Routineanalytik.
Graphit hat einen hohen elektrischen Widerstand und lässt
sich im Stromfluss bis zu 3000°C aufheizen. In einem Rohr aus Graphit lassen
sich deshalb ebenso wie in einer heißen chemischen Flamme Verdampfung und
anschließende Atomisierung einer Analysenlösung erreichen. Wird der
Lichtstrahl einer Strahlungsquelle auf die Atomwolke im Rohr gerichtet,
absorbieren die Atome des Analyten, für den die eingestrahlte Wellenlänge
spezifisch ist, entsprechend ihrer Konzentration Licht und die absorbierte Menge
liefert wieder die quantitative Information.
Der Sprung um rund drei Zehnerpotenzen in der Nachweisstärke
findet seine Erklärung in der höheren Verweilzeit der Atomwolke im Rohr im
Vergleich zur Flamme, der zwar ständiger Nachschub durch Ansaugung der Analytlösung
zur Verfügung steht, in der die Atomkonzentration aber dadurch nicht erhöht
wird, da ebenso ein stetiger Austrag aus der Flamme erfolgt. Aus dem Rohr, wenn
nicht durch Ausblasen gewollt, tritt die Atomwolke nur sehr langsam durch
Diffusion aus, Zeit genug, dass jedes Atom mehrfach vom unangeregten in den
angeregten Zustand wechseln kann und jedesmal seine Portion Licht absorbiert.
Anschaulich können also µg Analyt im Rohr ebenso viel Licht absorbieren wie mg
in der Flamme, in der andererseits µg keine Chance hätten, überhaupt messbare
Lichtschwächungen zu verursachen. Die Graphitrohr-AAS verdrängte die
Flammentechnik nicht, sondern ergänzte sie für niedrige
Konzentrationsbereiche. Höhere Konzentrationen im Rohr bedingen eine
Totalabsorption, ohne Verdünnung ist keine Messung mehr möglich und die
Flammen-AAS dann die geeignete Alternative.
Zwei spezielle Atomisierungstechniken, das Hydrid- und das
Kaltdampfverfahren, erlangten neben Flammen- und Graphitrohrtechnik in den
70er-Jahren Bedeutung und sind im modernen elementanalytischen Labor sogar oft
die einzigen Überlebenden der AAS-Aera. In beiden Fällen geht es um die auf
Grund ihrer chemischen Eigenschaften mögliche Abtrennung einzelner Elemente von
der restlichen Probenmatrix mit dem Vorteil, dadurch wesentlich ungestörtere
Messungen bei recht hoher Nachweisstärke verifizieren zu können.
Arsen (As), Selen (Se), Antimon (Sb) und wenige andere
Elemente bilden unter Reduktion mit Hydriden - in der Regel wird
Natriumborhydrid (NaBH4) eingesetzt - selbst flüchtige Hydride (AsH3
etc.), die mit Inertgas aus der Analysenlösung ausgeblasen und in einen
Atomisierungsraum eines Atomabsorptionsspektrometers, meist als heizbares
Quarzrohr ausgeführt, verbracht werden können. Dort werden sie bei rund 800°C
atomisiert, und die hydridbildenden Elemente auf ihren spezifischen
Absorptionswellenlängen vermessen.
Noch einfacher gelingt die Abtrennung des Quecksilbers (Hg)
aus der Analysenlösung. Auf Grund seines hohen Dampfdrucks lässt sich das
Element in reiner Form ausblasen, nachdem es durch Reduktion seiner Verbindungen
in der Analysenlösung erzeugt worden ist. Der Weg des Hg-Dampfes gleicht dem
der Hydride, indes erübrigt sich eine thermische Atomisierung, da der atomare
Zustand ja bereits erreicht ist. So findet die Absorption unter den ‚kältesten
Bedingungen’ aller AAS-tauglichen Elemente statt, was dem Verfahren seinen
Namen gab.
Das nicht nur ‚kälteste’, sondern auch nachweisstärkste
AAS-Verfahren aller Elemente wird daraus, wenn das Quecksilber vor der Messung
noch aufkonzentriert wird. Dies gelingt durch Amalgambildung an Metalloberflächen,
sehr geeignet ist Gold. Amalgame sind die Legierungen des Quecksilbers mit
Metallen, bilden sich mit Hg-Dampf rasch und lassen sich thermisch wieder
zersetzen. Ein zwischen Analysenlösung und AAS-Gerät platziertes Goldgewebe
von großer Oberfläche kann das Quecksilber auch mehrerer Analysenlösungen
anreichern und auf einen Schlag durch Erwärmung freisetzen, was natürlich im
Absorptionsbereich eine ungleich höhere Konzentration an Atomen ermöglicht,
als es allein durch das kontinuierliche Ausblasen aus der Analysenlösung
erreichbar wäre.
So lassen sich Nano- und Pikogrammmengen Quecksilber
nachweisen, vorausgesetzt ein Labor ist kontaminationstechnisch für
Ultraspurenanalysen eingerichtet. Ein übliches, gut ausgerüstetes
Spurenelementlabor im landwirtschaftlichen Untersuchungswesen wird sich mit 0,01
µg/l bescheiden und damit allen heutigen Anforderungen gewachsen sein.
|
|
|
|
Abb.29 Quecksilber-Kaltdampf-AAS |
|
Abb.29 zeigt das aktuell in Kassel seit 2001 eingesetzte
Kaltdampf-AAS mit kontinuierlicher Probenzuführung (Schlauchpumpen vorn),
Amalgamsystem (oben) und kontaminationsgeschütztem Probengeber (links).
Strahlungsquelle ist eine Quecksilberdampflampe, Absorptionsbereich ein
beheiztes Quarzrohr (hinter dem Sichtschlitz).
Innovation hat die Atomabsorptionsspektrophotometrie bis
heute reichlich erfahren: Kompensationstechniken für spektrale Interferenzen
(Untergrundkompensation), fortschreitende Automatisierung inclusive
PC-Steuerung, schnellere quasisimultane Strahlungsquellenrotation, Einsatz von
Kontinuum- statt elementspezifischer Strahlungsquellen, andere
Graphitrohrgeometrien, Feststoff- und Suspensionsverarbeitung in Graphitrohren,
kontinuierliche Hydridgenerierungs- und Probenzuführungssysteme, Kopplung der
Graphitrohr-AAS mit der Hydridtechnik und anderen Probenvorbereitungssystemen
u.v.a.m. und sie steht in der anorganischen Analytik wie keine zweite Technik für
den rasanten instrumentellen Wandel der Laborlandschaft der letzten 50 Jahre.
Trotzdem wurde sie als Folge der gesellschaftlichen und
politischen Anforderungen an die Labore des landwirtschaftlichen
Untersuchungswesens bis heute in vielen ihrer Anwendungen von noch schnelleren
und durchsatzstärkeren instrumentellen Messverfahren der Atomspektrometrie
abgelöst.
Den Anfang machte die ICP-Atomemissionsspektrophotometrie
(ICP-AES), die nach und nach der Flammen-AAS den Rang ablief, sich aber auch für
niedrigere Konzentrationsbereiche als diese einsetzen ließ. Es folgte die
ICP-Massenspektrometrie (ICP-MS), die sukzessive Aufgaben von der
Graphitrohr-AAS übernahm. Und schließlich erreichte die direkte
Elementanalytik aus der Festprobe eine neue Dimension mit labortauglichen Röntgenfluoreszensspektrometern.
Die Entwicklung einer durch elektromagnetische
Hochfrequenzinduktion erzeugten Plasmafackel (ICP = ‚inductive coupled
plasma’) als thermische Anregungsquelle bringt gemeinsam mit neuen Techniken
zur Herstellung leistungsfähiger Gittermono- und polychromatoren dem
Messprinzip Atomemission den lang erstrebten Durchbruch im elementanalytischen
Labor. Bei Temperaturen um 8000°C lässt sich eine Vielzahl von Elementen im
Gasplasma anregen. Um 1975 kommen die ersten Geräte auf dem Markt, das
bevorzugte ‚Brenngas’ ist Argon, es wird es bleiben. Noch sind die
Spektrometer in der Auswertung relativ langsam, weil bei komplexer
Probenzusammensetzung eine Vielzahl von Emissionslinien auszuwerten ist. Doch es
ist das Geburtsjahrzehnt des Microprozessors. Ende der 70er-Jahre hat er die
Spektrometrie erobert und revolutioniert die Mess- und Auswertegeschwindigkeiten
von Photometrie, AAS, AES etc. Ganz besonders aber profitiert die
Atomemissionsspektrophotometrie durch jetzt rechnergesteuerte Mono- oder
Polychromatoren. Solange sie mit einem Monochromator arbeitet, bleibt die AES
zwar ein sequentielles Verfahren wie die AAS, steuert jetzt allerdings die
Emissionslinien so schnell und präzise an, dass ein quasi simultaner Betrieb
resultiert, arbeitet sie gar mit einem Polychromator, der eine ganze Gruppe
parallel geschalteter Monochromatoren ersetzt, ist der Betrieb gänzlich
simultan.
Abb.30 zeigt das erste für die Anstalt in Kassel 1988
beschaffte ICP-AES, das bis 2007 noch seinen Dienst tat und auch mit einem
Polychromator ausgerüstet ist. Der abgebildete PC stammt allerdings aus den
90er-Jahren und repräsentiert schon die Enkel- oder Urenkelgeneration der
angesprochenen Mikroprozessoren.
|
|
|
|
Abb.30 ICP-Atomemissionsspektrometer |
|
Ob gänzlich oder nur quasi simultan, die Überlegenheit der
ICP-AES gegenüber der Flammen-AAS war rasch offensichtlich, Dutzende von
Elementen konnten nun gleichzeitig, mit geringeren spektralen Störungen und
ohne Einschränkung, ob geeignete Hohlkathoden- oder elektrodenlose
Entladungslampen verfügbar waren, bestimmt werden. Dennoch hat eine Fackel, ob
nun physikalisch oder chemisch erzeugt, die schon geschilderte Beschränkung,
dass angesaugter Analyt nicht lange verweilt, es damit auch für die ICP-AES
eine natürliche Grenze der Nachweisstärke gibt. Sie lässt sich mit besonderen
Techniken, etwa mit Ultraschallzerstäubung, zwar unter die der Flammen-AAS drücken,
jedoch nicht so niedrig, dass die ICP-AES auch Konkurrent der Graphitrohr-AAS hätte
werden können. Die ICP-Massenspektrometrie (ICP-MS) wurde es dann.
Mit der ICP-MS etabliert sich kommerziell ab 1985 eine
atomspektrometrische Technik, deren Messprozess nicht optisch ist. Atomisiert
und angeregt in der gleichen Art Plasmafackel wie wir sie gerade kennen lernten,
gelangt das Probenaerosol über ein Interface in ein Massenspektrometer. Dort
werden die Atome durch Elektronenbeschuss ionisiert, die Ionen als geladene
Teilchen in einem elektrischen Feld beschleunigt und durch elektrische Linsen
fokussiert, und der Ionenstrahl in ein Magnetfeld mit variierbarer Stärke
gelenkt. Ionen gleicher Masse und Ladung und deshalb gleicher Geschwindigkeit
werden abhängig von der Magnetfeldstärke gleichartig abgelenkt. Durch Veränderung
der Magnetfeldstärke ist es somit möglich, die verschiedenen Ionenarten
getrennt nach ihrer Masse auf einen ladungsregistrierenden Detektor zu lenken.
Die Masse charakterisiert das gefundene Element qualitativ, die gemessene
Gesamtladung aller dem Element zugehörigen Ionenladungen liefert im Vergleich
mit geeigneten Kalbrierproben die quantitative Information. Somit ist auch die
ICP-MS kein absolutes, sondern ein vergleichendes Messverfahren. Seine
Nachweisstärke gestattet es, noch Konzentrationen im Submikrogrammbereich
bezogen auf die Analytlösungen sicher zu analysieren. Grenzen hat die MS dort,
wo Massen unterschiedlicher Ionen gleich sind, störend hier insbesondere
solche, die sich durch Verbindungsbildung mit dem ‚Brenngas’ Argon bilden
(Argide). Seit einigen Jahren lässt sich hier mit sogenannten Reaktions- oder
Kollisionszellen Abhilfe schaffen, in denen Störionen durch Reaktion mit einem
Fremdgas am Eintritt in das Magnetfeld gehindert oder Analytionen (z.B. Arsen
(As+), Masse: 75) durch gezielte
Umsetzung mit einem Fremdgas (z.B. Sauerstoff (O2)) bei anderer Masse
(z.B. AsO+, Masse: 91) detektiert werden können. Die
Zellentechniken haben die Zahl der mit der ICP-MS erfassbaren Elemente deutlich
erhöht.
Röntgenfluoreszensspektrophotometrie (RFA) ist die jüngste
verfügbar gewordene instrumentelle Technik in der Routineelementanalytik.
Geeignete Spektrometer sind seit Mitte der 90er-Jahre im Handel. Die
bestimmbaren Konzentrationen reichen von wenigen bis zu einigen hundert mg/kg,
was andere vorgestellte atomspektrometrische Techniken auch leisten. Das
Besondere ist auch keineswegs die Nachweisstärke, sondern die Geschwindigkeit
der Bestimmung, da die chemische Probenvorbereitung entfällt, und die
Anwendbarkeit auf Elemente, die mit anderen atomspektrometrischen Methoden nicht
oder nur schlecht erfasst werden können, z.B. die Halogene.
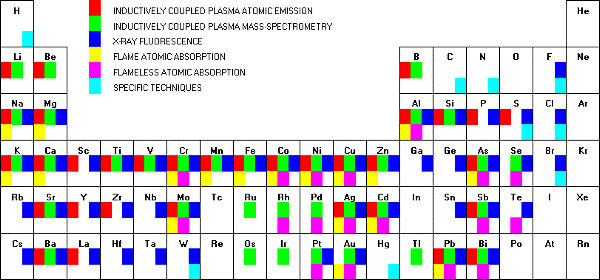
Abb.31 Geeignete atomspektrometrische Messtechniken
Schon aus der Graphitrohr-AAS war der direkte Einsatz von
Festproben geläufig. Breite Anwendung erfuhr diese Technik jedoch nicht, da nur
mit relativ geringen Mengen gearbeitet werden kann und mangelnde Probenhomogenität
dann größere Fehler bedingt. Eine bessere Homogenisierung ist jedoch oft so
aufwändig, dass alternativ auch gleich eine chemische Probenvorbereitung
(Aufschluss oder Extraktion) und Herstellung einer homogenen Analysenlösung
zeitlich konkurrieren kann. Das Homogenitätsproblem ist für die RFA wegen der größeren
einsetzbaren Probenmenge (bis zu mehreren Gramm) weit weniger existent, eine
hinreichende Vermahlung und Vermischung mit anschließender Komprimierung zu
einem Pressling aber für jede Probe erforderlich und Voraussetzung einer
korrekten Messung. Sie geschieht wieder nach einem optischen Prinzip. Der Probe
wird Röntgenstrahlung zugeführt, die sie absorbiert. Die aufgenommene Energie
verursacht verschiedene Elektronenprozesse (Anregung, Platztausch etc.) und wird
schließlich abhängig vom jeweiligen Prozess in unterschiedlicher Form wieder
emittiert. Ein Anteil, die sogenannte Fluoreszensstrahlung, wird für die
qualitative und quantitative Auswertung genutzt. Jedes Element hat spezifische
Fluoreszenswellenlängen.
Abb.31 verdeutlicht noch einmal farblich ohne Anspruch auf
Vollständigkeit, welche Elemente sich heute mit welchen der vorgestellten
atomspektrometrischen Methoden am besten bestimmen lassen.
Absicht des Kapitels sollte es nicht sein, den Leser mit
allen Detailentwicklungen der Atomspektrometrie der letzten 50 Jahren vertraut
zu machen, sondern für das folgende Kapitel eine gewisse Vorstellung zu
vermitteln, was die Analysensysteme zu leisten vermögen, die da unentwegt für
erkleckliche Barschaft beschafft werden, und für die fulminante instrumentelle
Revolution und Rationalisierung in der Elementanalytik gesorgt haben.
![]()
Das dritte halbe Jahrhundert
Es hat nach Vorstellung von soviel Messtechnik etwas tröstlich
Konservatives, dass im Laufe der 60er-Jahre in Kassel die Fluoranalytik in
Futtermitteln und Pflanzen entwickelt wurde, und wir vor den
atomspektrometrischen Zeiten noch ein Element in Augenschein nehmen können, das
sich dem neuen instrumentellen Ansturm noch nicht ergab, noch Anspruch auf eine
klassischere Behandlung anmeldete.
Auch den analytischen Chemikern in Darmstadt und Kassel wurde
in diesem Jahrzehnt deutlich, die goldenen Zeiten der Entdeckung, Entwicklung
und Anwendung hochspezifischer chemischer Reagenzien, zugegeben oft aufwendig,
aber immer spannend, neigten sich dem Ende zu. Physik und Technik eroberten das
Labor, und die chemischen Eigenschaften der Analyte traten gegenüber den
physikalischen in den Hintergrund. Ein neuer Industriezweig der Produktion
wissenschaftlich-technischer Geräte entstand, die analytische Chemie stellte
finanzielle Anforderungen in nie gekannter Höhe und die Analytiker erhielten
eine neue Verantwortung, die Angebote dieser Industrie fachlich und
kostenbewusst zu bewerten.
Nun, bei der Fluorbestimmung wurde noch destilliert und
titriert, der Grund der Fluoranalytik war aber ein ebenso neues Szenario wie es
die instrumentelle Aufrüstung darstellte, das wachsende Umweltbewusstsein und
der Wunsch nach Umweltschutz. Und eben jenes bzw. jener wurde in diesem
Jahrzehnt für die Elementanalytik wegweisend, erweiterte das Spektrum der
untersuchten Elemente und bedingte weiteren Bedarf an Automatisation und
Nachweisstärke. Es war ein z.T. sich gegenseitig bedingender Prozess, denn
manche Umweltproblematik verdankte ihre Entdeckung erst den neuen
Analysenautomaten.
Schauen wir zum Fluor (F). Fluorwasserstoff- (HF),
Siliciumtetrafluorid- (SiF4) und andere gasförmige Fluoridemissionen
sowie fluoridhaltige Stäube entwichen ungefiltert Aluminiumhütten, Ziegeleien,
Keramik- und Düngemittelfabriken und erhöhten in ihrer Umgebung die natürlichen
Gehalte in Pflanzenteilen von 0,5 - 10 mg/kg auf ein Vielfaches von bis zu 200 -
1000 mg/kg. Abwaschbar zeigte sich nur etwa die Hälfte des Fluors, die Pflanzen
entwickelten erhebliche phytotoxische Schäden. Um diese ursächlich zuordnen zu
können, diente für Pflanzen und Futtermittel folgende Methode. Unter Zusatz
von Quarz (SiO2) wurden die Fluoride mit Perchlorsäure
aufgeschlossen und als flüchtige Fluorkieselsäure (H2SiF6
od. SiF4 x 2 HF) per Wasserdampfdestillation von der restlichen
Matrix getrennt. Im Destillat erfolgte die Fluorbestimmung durch Titration mit
Thoriumnitrat (ThNO3) und Alizarinsulfonsäure als Indikator, die mit
Thorium einen roten Farbstoff bildet, wenn kein weiteres Thoriumfluorid (ThF4)
mehr gebildet wird.
1973 war die Fluoranalytik in Futtermitteln in Kassel
dann soweit ausgebaut, dass größere Serien gefahren werden konnten. So wurden
130 Heu- und Maissilageproben mit Gehalten zwischen 1,0 und 39,5 mg/kg
analysiert. Als unbedenklich galt ein Gehalt von <15 mg/kg, und in 95% der
Proben wurde diese Grenze nicht überschritten. Aber auch der Höchstgehalt lag
nicht mehr in den Dimensionen, wie sie noch wenige Jahre zuvor beklagt wurden,
ein deutliches Indiz dafür, dass sich zumindest im nordhessischen
Einzugsbereich der Anstalt die Kontaminationssituation durch Einsatz von
Filtertechnik durch die Industrie entspannt hatte. Da gleichzeitig Gefäßversuche
des Landwirtschaftlichen Versuchswesens belegten, dass der Transfer
Boden-Pflanze für Fluor eine untergeordnete Rolle spielte, wurden die
Fluorbestimmungen in Futtermitteln zur Kontrolle zwar noch etliche Jahre
weitergeführt, in den 80er-Jahren aber schließlich eingestellt.
Eine Umweltproblematik mit erheblich längerer
Lebensdauer und intensiverer Beachtung durch die Öffentlichkeit wurde Ende der
60er-Jahre dann zu einem wichtigen Argument für den Einstieg in die
Atomspektrometrie in Darmstadt und Kassel, die Bleibelastung durch den Straßenverkehr.
Als sogenanntes ‚Antiklopfmittel’ für Ottomotoren
mischte die Mineralölindustrie ihren Raffinaten das metallorganische
Bleitetraethyl (Pb(C2H5)4) in Konzentrationen
bis zu 500 mg Blei/l zu. Rund 50% davon wurde via Auspuffgase in die Umwelt
verbracht und machte den Begriff ‚Schwermetall’ in ganz anderer Weise, als
ihn die Chemie als ‚metallisches Element mit einem spezifischen Gewicht größer
5 g/cm3’ definiert, negativ publik. Da die Giftigkeit von Blei
vielen Menschen geläufig war, und sie nun das Element als Schwermetall
definiert bekamen, vermengten sich, wie auch bei Presse und Politik, die
Begriffe, und ‚Schwermetall’ wurde fortan meist ohne Ansehen der Dosis mit
‚Umweltgift’ und ‚gefährlichem langlebigem Schadstoff’ gleichgesetzt.
Dabei wirkte wissenschaftlich unterstützend, dass bei Blei keinerlei
essentielle Qualität als Spurenelement entdeckt wurde. Andererseits war
weitgehend unbekannt, dass zahlreiche essentielle Spurenelemente Schwermetalle
im chemischen Sinn waren und ihr segensreicher oder umweltbelastender Charakter
lediglich von der wirkenden Menge abhing. Als sich dann sogar Elemente, die der
chemischen Definition nicht entsprachen wie Arsen, Selen oder Antimon in der
Umweltdiskussion zum Umweltbegriff ‚Schwermetalle’ gesellten, führte dieser
in der Öffentlichkeit fortan ein von der chemischen Nomenklatur befreites
Eigenleben.
Wie wir schon erfahren haben, gab es nachweisliche
Schwermetallbestimmungen im mg/kg- oder mg/l-Maßstab im hessischen
landwirtschaftlichen Untersuchungswesen bereits 1900, und in den folgenden fünf
Jahrzehnten wurde immer wieder einmal eine kolorimetrische oder später
photometrische Schwermetallanalyse im speziellen Kontaminationsfall durchgeführt.
Ab den 50er-Jahren waren dann häufigere Schwermetallbestimmungen im Rahmen der
Spurenelementanalytik erfolgt, aber erst die Flammen-AAS ermöglichte die
Serienanalytik, nach der die neue Umweltdiskussion verlangte.
Das erste Flammen-AAS-Gerät schmückte 1968 das Kasseler
Labor, vorgesehen aber nicht für die Schwermetallanalytik, sondern zur
Abdeckung des stark gestiegenen Bedarfs an Magnesiumbestimmungen in Böden. Das
Erdalkalielement Magnesium ließ sich flammenphotometrisch nicht sehr gut
bestimmen, war hingegen so ideal mit der Flammen-AAS erfassbar, dass diese
Messung als einziges Flammen-AAS-Verfahren bis heute im hessischen
landwirtschaftlichen Untersuchungswesen überlebt hat (Abb.32). Um eine
Totalabsorption, wie oben für die Graphitrohrtechnik geschildert, zu
verhindern, muss, da es sich bei Magnesium ja nicht um ein Spurenelement
handelt, der für die spurenanalytische Anwendung konzipierte Absorptionsweg
durch die Flamme gemäß dem Lambert-Beerschen-Gesetz verkürzt werden, was
durch einfache Drehung des Brenners aus der Längs- (Abb.28) in die Querstellung
zum Lichtstrahl der Hohlkathodenlampe möglich ist, in Abb.32 an der schmalen
Flamme erkennbar.
Auf diesem AAS-Gerät gelangen aber auch die ersten
Gehversuche in der Bleianalytik. Bodenanalytik zumeist aus 1- oder 2-molarem
Salzsäureauszug im mg/kg-Bereich war direkt möglich, Pflanzenanalytik nach Säureaufschluss
im µg/kg-Bereich nach Anreicherung mit Dithizon.
Systematisch waren diese Gehversuche noch nicht. Die
ersten systematischen Untersuchungen unter Einbezug der als umweltschädigend
ebenso kritisch oder noch kritischer als Blei einzustufenden, wenn auch nicht
aus einer so ubiquitären Quelle wie dem Straßenverkehr stammenden
Schwermetalle Cadmium (Cd) und Quecksilber (Hg) erfolgten ab 1971 in Darmstadt
an Aufwuchs, nachdem auch dort die Flammen-AAS eingerichtet worden war.
Hauptquellen der Kontaminationen in der Landwirtschaft durch diese Schwermetalle
waren die Düngung und bei Quecksilber auch die Saatgutbeize.
|
|
|
|
Abb.32 Aktuelles
Flammen-AAS-Gerät für die Mg-Bestimmung |
|
Bevor wir die hinsichtlich der Schwermetallanalytik
illustren 70er- und 80er-Jahre Revue passieren lassen, sei noch ein
zusammenfassender Blick auf die 60er geworfen.
Das traditionelle Kerngeschäft der Elementanalytik, die
Mineralstoffanalytik an Düngemitteln, Futtermitteln und Böden lief weiter,
erlebte jedoch einen gewissen Rückgang an Bedeutung, da in einer wieder mit
Nahrungsmitteln versorgten und zu Wohlstand gekommenen Gesellschaft Raum für
andere Wünsche, aber auch Ängste entstand, was die Sicherung des Wohlstandes,
der gesunden Ernährung oder der Gesundheit allgemein anbetraf.
Die Elementanalytik erweiterte ihren Horizont durch die
neuen wissenschaftlichen Erkenntnisse über Mikronährstoffe bzw. essentielle
Spurenelemente und anorganische Schadstoffe, die, wie erläutert, nun gern unter
dem Begriff 'Schwermetalle' subsummiert wurden, beträchtlich und verfügte
gegen Ende des Jahrzehnts auch über die ersten Analysenautomaten, die in der
Lage waren, den reichlichen Hunger der Gesellschaft nach elementanalytischen
Daten zu befriedigen.
In Kassel widmete man sich z.B. nach vielversprechenden Düngungserfolgen
in Norddeutschland intensiv der Erforschung der Kupferversorgung hessischer Böden
und hessischen Aufwuchses. Rund 1200 Boden- und 500 Heuproben zeigten ein Bild
ausreichender Gehalte im Heu, aber z.T. unbefriedigender Versorgung der Böden.
Begleitet wurde das Programm bis 1975 von Feld- und Gefäßversuchen des
Landwirtschaftlichen Versuchswesens.
In Programmen um Spurenelemente sah sich die
Landwirtschaftliche Versuchsanstalt nun auch besonders als Mittlerin zwischen
Wissenschaft und Praxis. Es soll deshalb im weiteren Bericht auch nur mehr
sporadisch auf die Makroelemente 'NPK & Co.' geblickt werden, der Leser möge
sie aber, da sie auch heute noch einen hohen Untersuchungsanteil in der
Elementanalytik beanspruchen, bei den weiteren Schilderungen über
Spurenanalytik nicht als beendete Parameter empfinden.
Allerdings gingen die ehemals dominierenden Mineraldüngeruntersuchungen
mit der Einführung der Amtlichen Düngemittelkontrolle Ende der 60er-Jahre
nochmals zurück, eine Fortsetzung des Trends aus den 50ern und bedingt durch
die weiterhin verlässliche Produktion. Ein neues Arbeitsfeld mit
Schadstoffaspekten tauchte stattdessen im Zuge der angestrebten
landwirtschaftlichen Verwertung von Siedlungsabfällen auf.
Auf ein besonderes Kapitel Elementanalytik soll in dieser
Chronik nur verwiesen werden, da es in einem Band der in den 90er-Jahren
verlegten Schriftenreihe der Hessischen Landwirtschaftlichen Versuchsanstalt
hinreichend beschrieben ist, das Messwesen radioaktiver Isotope in Umweltproben.
Ausgelöst durch den Fallout der Kernwaffenversuche der 50- und 60er-Jahre wurde
auch die Versuchsanstalt ab 1962 in das nationale Überwachungsprogramm
eingebunden. Sie leistete in einem eigens eingerichteten Dezernat in Darmstadt
zunächst nur die aufwändige Probenaufarbeitung und ließ die Messungen außerhäusig
durchführen, stieg aber 1972 im Rahmen der Umgebungsüberwachung des hessischen
Kernkraftwerkes Biblis selbst in das Messwesen ein. Große Bedeutung erlangten
die Radioaktivitätsmessungen 1986 und dessen Folgejahren nach dem tragischen
Kernkraftwerksunfall in Tschernobyl. Im Jahr 2000 wurde dieser Aufgabenbereich
der Elementanalytik an eine andere hessische Behörde verlagert.
Die 60er-Jahre endeten mit dem 'Gesetz über die Auflösung
der Land- und Forstwirtschaftskammern Hessen-Nassau und Kurhessen', und 1970
nach über 100 Jahren resultierte da-raus die Vereinigung beider Anstalten unter
der Regie der hessischen Agrarverwaltung und der gemeinsame Name 'Hessische
Landwirtschaftliche Versuchsanstalt' (HLVA).
Für die Element-, insbesondere die Mikroelementanalytik
mit Umwelthintergrund hatte dies in den Folgejahren die Konsequenz einer Art
Aufgabenteilung zwischen Kassel und Darmstadt, die zwar nicht alle bestehenden
Parallelarbeiten aufhob, aber teilweise schon vorhandenen Schwerpunkten folgend
in Darmstadt mehr die Bearbeitung landwirtschaftlicher Produkte (Aufwuchs,
Fleisch, Fisch etc.) und in Kassel mehr die Bearbeitung landwirtschaftlicher
Produktionsmittel (Boden, Düngemittel, Siedlungsabfälle, wirtschaftseigene
Futtermittel etc.) und von Proben aus dem Landwirtschaftlichen Versuchswesen
konzentrierte. Bei den Handelsfuttermitteln blieb es bei der Bearbeitung an
beiden Standorten.
Die 70er- und 80er-Jahre brachten der Elementanalytik vor
dem Hintergrund neuer gesellschaftlicher Standpunkte zu Qualität und
Schadstofffreiheit landwirtschaftlicher Produkte und Produktionsgrundlagen einen
ungeahnten Aufschwung. Die Anstalt wurde dem gerecht, indem sie 1976 in
Darmstadt und 1980 in Kassel zwei neue Dezernate für Aufgaben in der Element-
und Umweltanalytik einrichtete und zwei weitere Chemiker an Bord nahm. 1978
wurde auch dem starken Zuwachs der elementanalytischen Arbeiten in der Analytik
von Siedlungsabfällen mit der Errichtung neuer Laboratorien in Kassel Rechnung
getragen.
Das zuständige Ministerium stellte der Elementanalytik für
die erforderliche Ausstattung mit zunächst Atomabsorptionstechnologie, später
anderen atomspektrometrischen u.w. Analysen- und Probenaufschlusssystemen Mittel
in bisher nicht gekanntem Umfang zur Verfügung. In den 80ern beteiligten sich
dann auch die Forstbehörden aus noch zu beschreibenden Entwicklungen heraus an
der instrumentellen Aufrüstung.
Nach dem Selbstverständnis landwirtschaftlicher
Untersuchungsanstalten waren sie schon immer mit dem Schutz der Umwelt betraut
und betrachteten es als konsequent, dass politischer Wille sie im 'Überwachungsbereich
Landwirtschaft' einsetzte. In der gesellschaftlichen Bewertung war der Stand der
Landwirtschaft und der u.a. für ihr Wohl arbeitenden Untersuchungsanstalten
keineswegs so eindeutig auf der Seite der 'Guten', und es bedurfte einer
umfangreichen Leistung in der Umweltanalytik, den Ruf der Anstalten als Garanten
von Nahrungsqualität und Schadstofffreiheit zu manifestieren.
Die Elementanalytik tat das ihre dazu. Seit 1973 sind für
die vereinte Anstalt wieder durchgängig Jahresberichte verfügbar, die das mit
Untersuchungszahlen und auch Berichten über Schwermetallvegetationsversuche
belegen. Der Chronist kann daraus nur eine Auswahl präsentieren, eine auch nur
annähernde Darstellung des elementanalytischen Untersuchungss-umfangs der
letzten 35 Jahre wäre zwar imposant, aber doch wohl ermüdend.
Schon 1973 wurden in rund 5% der Bodenproben (ca. 25.000)
ein oder mehrere Schwermetalle bestimmt, teils mit der Fragestellung
Spurenelementversorgung, teils mit der Fragestellung Schadstoffbelastung, ein
Prozentsatz, der nach einem Hoch von rund 10% in den 80er-Jahren, auch heute
wieder gilt, allerdings für die doppelte Anzahl Proben. Wohlgemerkt ist dies
eine Ausssage für die Schwermetall- und nicht die Elementanalytik, die fast
jede Bodenprobe zählen dürfte.
Rund 1000 Schwermetallanalysen steuerte im gleichen Jahr
auch schon die Futtermittelanalytik bei, thematisch noch ganz auf
Spurenelementsuche. Systematische Arsen-, Blei-, Cadmium- und
Quecksilberanalysen an Futtermitteln, den vier unerwünschten Schadstoffen mit
Elementcharakter in der heutigen Futtermittelverordnung, wurden dann ab 1977 in
Darmstadt in die Routine aufgenommen.
1974 wurde die elementanalytische Geschichte der
Versuchsanstalt durch umfangreiche Gefäßversuche zur Blei-, Cadmium- und
Quecksilberaufnahme bereichert, wobei Versuche an beiden Standorten angelegt
wurden, Quecksilber aber eine Spezialität Darmstadts war. Insbesondere galt das
Interesse dem Transferverhalten Boden-Pflanze des toxikologisch bedenklichen
Methylquecksilbers. 1974 war auch das Jahr, in dem die Atomspektrometer an
beiden Standorten mit Hydrid- und Kaltdampftechnik nachgerüstet wurden.
1975 sah den Beginn der ersten Vegetationsversuche mit Klärschlamm
bei besonderem Augenmerk auf Transferfragen von Blei und Cadmium sowie die
ersten Schadstoffreihenuntersuchungen an Klärschlämmen und Müllkomposten in
Kassel. Analyten waren neben Blei und Cadmium Arsen, Chrom, Kupfer, Mangan,
Nickel, Quecksilber und Zink.
1976 wurde in Darmstadt ein langjähriges
Untersuchungsprojekt zum Quecksilbermonitoring von Rhein- und Mainfischen
aufgelegt, das 1980 um die Elemente Blei und Cadmium erweitert wurde und bei
seiner Auswertung 1983 rund 900 Quecksilber- und 250 Blei- und Cadmiumproben
umfasste. Auslöser war eine gesetzliche Verordnung von 1975, die erstmals eine
Höchstmenge für Quecksilber in Fischen und anderen marinen Organismen
festlegte. In guter Tradition zu früheren Darmstädter Methodenentwicklungen
bei Probenaufschlüssen (s.o.) wurden verschiedene Aufschlussvarianten
entwickelt. Neben einem nur für die Hg-Bestimmung eingesetzten Salpetersäure-Schwefelsäure-Aufschluss
im offenen System, bei dem unter Rückfluss und Salpetersäurezuspeisung nahe
dem Siede- und Zersetzungpunkt der Schwefelsäure von 340°C aufgeschlossen
wurde, wurde noch ein für alle drei Elemente anwendbarer Salpetersäure-Wasserstoffperoxyd-Druckaufschluss
(Abb.33) mit einer maximalen Aufschlusstemperatur von 160°C und ein nur für
die Cd- und Pb-Bestimmung herangezogener Salpetersäure-Perchlorsäure-Aufschluss
mit einer maximalen Aufschlusstemperatur von 220°C geprüft.
|
|
|
|
Abb.33 Druckaufschlussbomben 1985 |
|
Der offene Hochtemperatur- und der Druckaufschluss
erwiesen sich im Zusammenhang mit der Kaltdampf-AAS als geeignete Varianten für
die Hg-Bestimmung in Fischen. Erfreulicherweise war im Untersuchungszeitraum
auch ein stetiger Rückgang des Belastungsgrades zu verzeichnen.
Bei der Blei- und Cadmiumbestimmung konnte der
Druckaufschluss zunächst nicht überzeugen. Da unter den gewählten Bedingungen
zwar, wie ein Vergleich mit der Perchlorsäurevariante belegte, die
Schwermetallverbindungen aufgeschossen wurden, die ganze Probe jedoch insgesamt
schlechter, waren die Graphitrohr-AAS-Messungen - über die Technik verfügte
die HLVA seit 1977 - stark untergrundgestört. Erst die technische Innovation
der sogenannten Zeeman-Untergrundkompensation (Abb.34)
für die
Graphitrohrtechnik und ihre kommerzielle Verfügbarkeit ab 1980 machten dann
den immer mit der Hypothek der Explosionsgefahr behafteten Perchlorsäure-Aufschluss
obsolet und den Druckaufschluss für Proben mit vorwiegend organischer Matrix
hoffähig.
Und zwar so hoffähig, dass auf der Basis der Darmstädter
Ergebnisse dieser Aufschluss Anfang der 80er-Jahre als Verbandsmethode des
VDLUFA in das Methodenbuch der 1974 gegründeten Fachgruppe ‚Umweltanalytik’
aufgenommen wurde. Die Fachgruppe ‚Umweltanalytik’ wurde die Heimat der
Spurenanalytiker der HLVA und wechselseitig beeinflusste man sich stark, bis
schließlich der Fischanalytiker von eben, unser geschätzter E.Janßen, in den
90ern gar ihr langjähriger Vorsitzender wurde.
|
|
|
|
Abb.34 Graphitrohrofen mit Zeeman-Untergrundkompensation 1985 |
|
Die Jahre 1978-85 waren in Politik, Gesellschaft und
Wissenschaft von einer starken Hinwendung zur Schadstoffproblematik
‚Schwermetalle’ geprägt, die mitunter sogar Züge der Hysterie trug. Die
HLVA profitierte davon in Form einiger öffentlicher Forschungsaufträge. Wie
wichtig das Forschungsgebiet auch im VDLUFA gesehen wurde, demonstrierte das
Generalthema 'Schwermetalle in der Nahrungskette - Belastungsgrenzen für
Mensch, Tier und Pflanze ' des Kongresses 1982 in Münster.
Vorbereitungsphase, Inkraftsetzung 1983 und Vollzug der
ersten deutschen Klärschlammverordnung (KSVO) mit Schwermetallgrenzwerten für
Klärschlämme und Böden vervielfachte in kurzer Zeit die Zahl der
Schwermetalluntersuchungen insbesondere bei Böden. Die wissenschaftlich
begleitete Festlegung von justitiablen Bodengrenzwerten wirkte weit über die
Verordnung hinaus, und die Werte fanden vergleichende Anwendung bei einer ganzen
Reihe von hessischen Bodenmonitoringprogrammen.
Bodenanalysen von landwirtschaftlichen Flächen nahe
Autobahnen waren 1978 eines davon. Bereits Mitte der 60er-Jahre hatte A.Kloke
den Verzicht des Anbaus von Nahrungs und Futterpflanzen in größerer Nähe zu
stark befahrenen Straßen gefordert, solange Bleizusätze zum Benzin gesetzlich
erlaubt blieben. Seine systematischen Pflanzen- und Bodenuntersuchungen zur
Thematik, die ersten in Deutschland, führte er noch ohne AAS durch und
entwickelte eigens ein kolorimetrisches Bleidithizonatverfahren. Da hatten wir
Hessen es ein Jahrzehnt später mit allen Spielarten der AAS ausgestattet
erheblich leichter. Im wesentlichen wurden die Kloke’schen Erkenntnisse bestätigt,
die hoch belasteten Kontaminationszonen endeten wenige Meter neben den Straßen,
bis zum Abstand von 50-100 m waren aber noch Belastungen messbar. Begleitende
Pflanzenanalysen und Vegetationsversuche zeigten die teilweise Abwaschbarkeit
von Blei und seine gegenüber anderen Schwermetallen vergleichsweise schlechte
Pflanzenverfügbarkeit.
1979-81 folgten den Autobahnflächen statistische
Bodenbeprobungen von Acker-, Grünland- und Weinbergsflächen sowie aus mutmaßlich
belasteten und unbelasteten städtischen Kleingartenanlagen. Insgesamt wurden in
Kassel über 3000 Böden auf ihre Gehalte an Blei, Cadmium, Chrom, Kupfer,
Nickel, Quecksilber, Zink, die 7 Elemente der KSVO, und Arsen untersucht,
darunter 2200 Ackerböden aus der ‚Besonderen Ernteermittlung (BEE)’, einer
amtlichen Probenahme, die der jährlichen Überprüfung des Nährstoffstatus
hessischer Ackerflächen und der
Ernteertragsermittlung diente. Um zu grenzwertvergleichbaren mittleren
Schwermetallgesamtgehalten zu gelangen, wurden rund 10% der Proben einem
Schwefelsäure-Salpetersäure-Aufschluss unterworfen, sodann alle Proben mit 2
molarer Salzsäure extrahiert, und die extrahierbaren Gehalte mit den Faktoren
multipliziert, die sich aus dem Vergleich Gesamtgehalt zu extrahierbarem Gehalt
der 10% beiden Methoden unterworfenen Proben im Mittel ergeben hatten. Daraus
formte sich ein Bild relativ unbelasteter hessischer Ackerflächen. Es wurde
1983 abgesichert, als von weiteren 1000 Ackerböden aus der ‚BEE’
Gesamtgehalte aus Königswasseraufschlüssen erhalten wurden, die mit den
errechneten gut korrelierten.
|
|
|
|
Abb.35 Rückflussbatterie für Königswasseraufschlüsse |
|
Der Königswasseraufschluss (Abb.35), wie ihn die KSVO
beschreibt, hat sich seit 1983 als Standardnassaufschluss der HLVA für Matrices
vorwiegend anorganischer Zusammensetzung bewährt. Er liefert annähernde
Gesamtgehalte, da Silikate und einige Oxide mit möglichen Schwermetallanteilen
nicht aufschließbar sind. Die Anteile gelten jedoch als nicht umweltrelevant.
Ein großes Plus des Aufschlusses ist seine gute Reproduzierbarkeit.
Der Standardaufschluss für Matrices vorwiegend organischer
Zusammensetzung war über Jahrzehnte allein die trockene Veraschung im
Muffelofen. Inzwischen war der Druckaufschluss als Nassaufschluss hinzugekommen.
Ab 1980 wurde in Kassel mit einer weitereren trockenen Variante experimentiert,
die auf Grund ihrer hochstehenden Technik und Investition eine Erwähnung
verdient hat, auch wenn sie sich letztlich als zu akademisch und durch zu
geringen Probendurchsatz als ungeeignet für die Routineanalytik erwies. Die
Rede ist von der Vakuumtieftemperaturveraschung im angeregten Sauerstoffplasma,
kurz: Plasmaveraschung (Abb.36).
|
|
|
|
Abb.36 Plasmaverascher 1980 |
|
Vakuum hieß hier Druck um 1 mbar, Tieftemperatur im
Vergleich zur wesentlich heißeren Muffelveraschung eine Maximaltemperatur bis
200°C, thermisch also ein sanfter Aufschluss für empfindliche Proben, gedacht
auch, Verluste leichtflüchtiger Schwermetalle auszuschließen, der sein
Aufschließvermögen nicht aus der Hitze sondern aus der Oxidationskraft des
reinen und hochreaktiven angeregten Sauerstoffs bezog. Die Vollständigkeit der
Aufschlüsse wurde in Arbeiten mit Klärschlämmen und Aufwuchs nachgewiesen,
indes waren die Veraschungszeiten für ein Standardverfahren nicht tolerabel. Investiert wurde zu Beginn der 80er-Jahre auch in zur
optischen Atomspektrometrie alternative Messtechniken. So wurde 1981 in Kassel
ein elektrochemischer Messplatz für Polarographie und Inversvoltammetrie
(Abb.37) eingerichtet. Mit den beiden verwandten elektrolytischen Techniken
konnte Schwermetallanalytik im mg/kg- und µg/kg-Bereich betrieben werden.
|
|
|
|
Abb.37 Polarographischer Messplatz 1981 |
|
Während in Darmstadt die Graphitrohr-AAS seit 1978 intensiv
eingesetzt und erforscht wurde, war sie in Kassel probenartbedingt selten in
Gebrauch und geriet ab 1981 durch den Einsatz der Inversvoltammetrie fast ins
Abseits, da mit dem elektrochemischen Verfahren der aktuelle Probenanfall im µg/kg-Bereich
hinreichend bearbeitet werden konnte.
Als die HLVA allerdings 1984 das Darmstädter
Schwermetalldezernat samt seinem hohen Probenaufkommen nach Kassel verlegte und
mit ihm das leistungsstarke, methodenvalidierte Graphitrohr-Equipment, setzte
sich die Atomspektrometrie schnell und eindeutig als Methode der Wahl durch, da
sie einen wesentlich höheren Probendurchsatz garantierte. Die elektrochemischen
Verfahren blieben noch bis 1990 als Referenzverfahren im Einsatz.
Der wohl langfristig für die Elementanalytik bedeutendste
Auftrag aus den Jahren 1978-85 erwuchs aus der Zusammenarbeit der Anstalt mit
den hessischen Forstbehörden. Die HLVA übernahm 1979 quasi die Funktion eines
hessischen Forstlabors und war fortan am Kampf der ‚Hessischen Forstlichen
Versuchsanstalt’ gegen die sogenannten ‚neuartigen Waldschäden’
beteiligt. Anfangs hieß das umfangreiche Programm, das das Land Hessen mit der
Einrichtung zahlreicher Messstationen in hessischen Wäldern und der
Untersuchung abertausender Boden-, Pflanzen-, Wasser-, Staub- und Luftproben
manifestierte, 'Waldschäden durch Immissionen', in den 90er-Jahren wurde es in
'Waldökosystemstudie Hessen' umbenannt, und es gesellten sich die
Waldschadensaktivitäten der Europäischen Union als
'EU-Level I'- und 'EU-Level II'-Programm hinzu. Zu Anfang waren die
Schwermetalle in Verdacht, eine der Hauptursachen der Waldschäden zu sein, später
setzte sich dann eine mehrfaktorielle Sichtweise durch, die auch biologische
Einflüsse berücksichtigte
Für das nach Kassel übersiedelte Dezernat, das im Laufe der
80er-Jahre mit der forstlichen Pflanzen- und Wasseranalytik den Löwenanteil der
forstlichen Untersuchungen zu leisten hatte, wurden 1984 und 1985 umfangreiche
Investitionen in der Atomspektrometrie getätigt. 3 weitere Graphitrohr-AAS-Geräte
mit Zeeman-Untergrundkompensation und ein zentrales Steuerungs- und
Datenverarbeitungssystem machten das Messlabor zu einem der schlagkräftigsten
elementanalytischen Labore im VDLUFA. Abb.38 lässt uns einen Blick in das neu
eingerichtete Labor werfen.
Auch das angestammte elementanalytische Dezernat, das die
forstliche Boden- und Staubanalytik per Flammen-AAS bearbeitete, durfte sich über
Investitionen bei Gasversorgung, Abluftanlage, Abzugstechnik für Königswasseraufschlüsse
und Datenverarbeitungsystemen freuen.
|
|
|
|
Abb.38 Graphitrohr-AAS-Labor 1985 |
|
Die umfangreiche elementanalytische Aufrüstungsphase traf
sich im übrigen kongenial mit dem Erscheinen und den Möglichkeiten des
Personalcomputers. Der Automatisierung der Analysengeräte diente der
Mikroprocessor bereits seit einem Jahrzehnt. Nun veränderte sich sein Wirken
von interner zu externer Steuerung. Gleichzeitig nahm die Vielfalt der Software
zu. Diese Entwicklung bei der AAS und der Kauf des ersten
ICP-Atomemissisonspektrometers 1988 (Abb.30) ermöglichte einen vorher nicht für
möglich gehaltenen Durchsatz, der, die Elementeinzelanalysen beider Dezernate
zusammengerechnet, zum Ende des Jahrzehnts nach Hunderttausenden zählte.
1984 konnte die HLVA nicht zuletzt aus der Entwicklung der
Umwelt- und Spurenanalytik heraus die vorgesetzten Dienststellen von der
Notwendigkeit eines Laborneubaus in Kassel überzeugen. Nach dreijähriger
Planungsphase unter intensiver Berücksichtigung des analytischen Knowhows der
HLVA wurde das neue, technisch hochgerüstete Laborgebäude ab 1987 vom
Hessischen Staatbauamt errichtet und 1991 übergeben. Bis 1997 folgten noch
Modernisierungen, die das alte Gebäude an den technischen Stand des Neubaus
anpassten.
|
|
|
|
Abb.39
Mikrowellenbeheiztes |
Abb.40
Hochdruckaufschluss- |
Neben dem raumtechnischen Quantensprung war es der
Elementanalytik 1991 auch noch einmal vergönnt, großzügige Geräteinvestitionen
aus dem Bautitel zu erhalten, die in die Beschaffung verschiedener
instrumenteller Aufschlussapparaturen (Abb.39/40), Elementaranalysatoren
(Abb.41/42) und des ersten ICP-Massenspektrometers flossen. Nicht ohne Stolz
verwiesen die Dezernenten der beiden Dezernate Anfang der 90er-Jahre auf ihren
gemeinsamen Gerätepark im Beschaffungswert von rund 3 Mio. DM.
|
|
|
|
Abb.41 Kohlenstoffanalysator 1991 |
Abb.42 Stickstoffanalysator 1992 |
Der mikrowellenbeheizte Druckaufschluss
(Abb.39) löste den
oben gezeigten elektrothermisch beheizten Druckaufschluss in Bomben (Abb.33) bei
ansonsten wenig veränderten Aufschlussbedingungen ab, der Hochdruckaufschluss
(Abb.40) verkörperte das gleiche Aufschlussprinzip, ermöglichte aber
wesentlich höhere Aufschlusstemperaturen über 300°C, die sich als
erforderlich für die Zerstörung bestimmter element-organischer Bindungen
herausgestellt hatten, z.B. solcher des Arsens in marinen Futtermitteln.
Mit den Elementaranalysatoren zur C-
(Abb.41) und
N-Bestimmung (Abb.42) kehrte die uralte Dumas'sche Verbrennungsanalytik (s.o.)
in die HLVA zurück und löste nasschemisch-oxidative C- und
Kjeldahl-N-Bestimmungen hauptsächlich in der Bodenanalytik ab. Beide Geräte
veraschten die Proben in reinem Sauerstoff und bestimmten die
Verbrennungsprodukte Kohlendioxid (CO2) bzw. Stickstoffdioxid (NO2)
infrarotspektroskopisch bzw. über die Veränderung der Wärmeleitfähigkeit
eines Trägergasstromes. Vor über 100 Jahren hießen die Bestimmungsverfahren
Gravimetrie bzw. Gasvolumetrie, die Verbrennung und Aufreinigung der flüchtigen
Verbrennungprodukte folgte aber damals schon dem gleichen Prinzip wie heute.
Die ICP-MS wurde ab 1993 mehr und mehr Arbeitspferd der
Spurenanalytik und ersetzte sukzessive die Graphitrohr-AAS, die es aber für
bestimmte Elemente wie Eisen, Chrom und Nickel, die in der Massenspektrometrie
interferierenden Störungen unterworfen waren, nach wie vor und als
Referenzverfahren brauchte. Das Schwermetallmesswesen beider elementanalytisch tätiger
Dezernate verlagerte sich nun durch die moderne ICP-Ausrüstung immer mehr auf
das von Darmstadt gekommene Dezernat.
Trotz in den 90er-Jahren weiterhin und durchaus stark
steigender Probenzahlen, die sich zum Großteil aus weiter laufenden amtlichen
Aufträgen speisten, neigte sich in diesem Jahrzehnt die Aera der politisch überproportional
beachteten anorganischen Schadstoffanalytik sanft dem Ende zu. Ein Ereignis, das
schon schlagartig die öffentliche Sichtweise über Umweltbedrohungen veränderte,
war dabei sicher die Reaktorkatastrophe von Tschernobyl 1986.
Auch war inzwischen klar, dass Schwermetalle nicht die
Hauptverursacher der forstlichen Schäden waren und das diesbezügliche
Aufkommen an Aufwuchs- und Bodenproben ließ nach. Dem voraus gegangen war
allerdings 1992/93 als Sonderprogramm noch die 1. bundesweite forstliche
Bodenzustandserhebung (BZE I) mit rund 900 Böden von 140 hessischen
Rasterstandorten und rund 30.000 Einzelelementuntersuchungen, die ganz aktuell in 2007
als Trendstudie 'BZE II' mit den gleichen Beprobungspunkten wiederholt wird.
Weiterhin unvermindert blieb zunächst das Interesse der forstlichen Forschung
an der Hydrologie. Jährlich 5.000 bis 6.000 forstliche Wasserproben mit 100.000
-120.000 Einzelelementanalysen sorgten mit für eine nimmermüde ICP-AES und
ICP-MS.
1997 übernahm E.Janßen die Leitung der Anstalt, und die
beiden Dezernate wurden unter der Leitung des Chronisten vereint.
Etwa Mitte der 90er-Jahre begann eine Zeit, die bis heute anhält,
die von leereren öffentlichen Kassen, Personalabbau, Aufgabenkritiken,
Umorganisationen und bürokratischen Auflagen geprägt wird. Das hatte auch
Konsequenzen für die Schlagkraft der Elementanalytik. Relativ zufrieden mit den
verbliebenen investiven Möglichkeiten (s. Abb.43/44/45) und mit gewissem Glück
bei der Altersstruktur der MitarbeiterInnen in der Elementanalytik auch von der
Personalreduzierung weniger betroffen als andere Bereiche - wobei die Schließung
der Anstalt in Darmstadt 1999 mit lediglich torsoartiger Verlegung der Reste
nach Kassel hier wohl das augenfälligste Ereignis war -, wurde jedoch die verfügbare
Zeit zu innovativer und kreativer Beschäftigung mit der Materie Spuren- und
Elementanalytik - es darf auch Angewandte Forschung dazu gesagt werden - durch
vielfältige von Politik und Verwaltung hereingetragene Beschäftigungszwänge
so eingeengt, dass nicht viel mehr als die ‚übliche Routine' übrig blieb und
mit gewisser Sehnsucht auf die Jahrzehnte geblickt werden muss, in denen in der
Elementanalytik wissenschaftlich etwas bewegt werden konnte und auch aus
vorgesetzten Dienststellen fachliches Verständnis und nicht nur Kostenanfragen
kamen.
|
|
|
|
Abb.43
ICP-Atomemissionspektrometer |
Abb.44
Mikrowellenbeheiztes |
|
|
|
Abb.45
ICP-Massenspektrometer mit |
Zu den Beschäftigungszwängen zählt trotz seiner die
Analytik durchaus befruchtenden Teile auch das Qualitätsmanagement, da es sich
fort von der Befruchtung, die bei der erfolgreichen Akkreditierung der HLVA 1998
nach der damaligen Norm DIN 45001 noch spürbar war, mit der DIN EN ISO 17025
doch zu einem Dokumentenmoloch mauserte. Die Elementanalytik ist mit über 70 Prüfverfahren
akkreditiert und steuert dementsprechend fleißig Dokumente bei. Hochwertige
Qualitätssicherung war auch vor der Akkreditierung schon Maxime, wie
jahrzehntelange, umfangreiche Teilnahmen an Ringuntersuchungen und
Methodenentwicklungen belegen können. Nur sind Qualitätssicherung und Qualitätsmanagement
leider oft zweierlei Ding. Der hier Kritik übt, hat das Haus mit gewissem Stolz
in die Akkreditierung geführt und fungiert auch heute noch als sein Qualitätsmanagementbeauftragter,
eigentlich also ein Motivierter.
Die skeptische Sichtweise wäre möglicherweise gedämpfter,
wenn das Qualitätsmanagement der einzige das Fachliche beschneidende Faktor
geblieben wäre.
Doch zu schultern waren und sind Privatisierungsplanspiele
Ende der 90er, zwei betriebliche Umorganisationen infolge Einbindung in das
neugegründete Hessische Dienstleistungszentrum für Landwirtschaft, Gartenbau
und Naturschutz (HDLGN) 2001 und 2005 bereits wieder in
den neugegründeten Landesbetrieb Hessisches Landeslabor (LHL) mit jeweils
umfangreichen organisatorischen und betriebswirtschaftlichen Veränderungen, die
Einführung einer betriebswirtschaftlichen Software (SAP) 2001 und deren
Neuanpassung 2005 und eine die gesamte bestehende fachliche Datenbankstruktur
verändernde Einführung eines neuen Labor- und Informationsmanagementsystems
(LIMS) 2007, um nur die wichtigsten zu nennen.
Das erste Jahrzehnt des neuen Jahrhunderts, das 15. der
Anstalt, bot und bietet an Nichtfachlichem also einige Abwechslung.
Doch lassen wir das dritte halbe Jahrhundert mit
Fachlichem ausklingen.
Von 2000 bis zur Eingliederung der Anstalt in das LHL wurden
die in Tabelle Abb.46 gelisteten jährlichen Untersuchungsleistungen erbracht.
|
Jahr |
2000 |
2001 |
2002 |
2003 |
2004 |
|
Messungen |
191.000 |
185.000 |
193.000 |
190.000 |
165.000 |
|
Abb.46 Zahl der Elementbestimmungen 2000 - 2004 |
|||||
Das sind stolze Zahlen. In die Summen sind keine
Mehrfachbestimmungen und Messungen des Qualitätsmanagements
(Ringuntersuchungen, Gerätekalibrierungen, Validierungen) eingerechnet. Das
gilt auch für 2003, in dem die erfolgreiche Reakkreditierung der Anstalt gelang.
Mit dem Rückgang der Analysen 2004 kündigte sich der
Ausstieg der hessischen Forstwirtschaft aus der 26 Jahre währenden Liaison mit
der Anstalt an. Die Gründung einer forstwissenschaftlichen Einrichtung durch
mehrere Bundesländer, darunter auch Hessen, die ihr Labor außerhalb Hessens wählte,
beendete 2005 mit Ausnahme der noch laufenden Arbeiten an der BZE II die langjährige,
fruchtbare und für die Elementanalytik beschäftigungsreiche Zusammenarbeit.
Der Verlust der Forstproben reduzierte die Probenzahl um rund 1/5 und die
Einzelelementbestimmungen um rund 1/4.
Einen Ausgleich, nicht ganz an Menge, aber doch an Vielfalt
der Probenarten und neuen Untersuchungsmethoden bot ab 2006 die Übernahme der
Elementanalytik für Lebensmittel, Bedarfsgegenstände und Kosmetika im gesamten
neuen Landesbetrieb. In Marburg und Kassel-Harleshausen war die
Lebensmittelanalytik schon einmal vom ausgehenden 19.Jahrhundert bis 1957
beheimatet gewesen und mit der neuen Zuständigkeit kehrte so etwas wie ein Stück
vom verlorenen Kinde heim.
So endete das Untersuchungsjahr 2006 mit einer
Gesamtprobenzahl von rund 20.000 bei rund 140.000 Einzelelementbestimmungen.
Das zeigt auch, dass Nichtfachliches noch nicht die Oberhand
gewonnen hat. So konnten wir uns auch in den letzten Jahren über schöne
analytische Erfolge freuen, so z.B. in der Selen- und Arsenanalytik mit der
Eigenentwicklung einer speziellen Hydrid-Graphitrohr-AAS-Kopplung für die
Selenbestimmung und einer methodischen Entwicklung für die Arsenbestimmung, die
die Möglichkeiten des mikrowellenbeheizten Hochdruckaufschlusses (Abb.44) und
der Kollisionszellentechnik der neuen ICP-MS (Abb.45) kombinierte. Die letzte
apparative Errungenschaft war 2006 ein Röntgenfluoreszenzspektrometer, das nun
erfolgreich in der Futtermittelelementanalytik seinen Dienst tut.
Das dritte halbe Jahrhundert veränderte die Laborlandschaft
entscheidend, es machte sie instrumentell und digital, leistungsfähig, aber
auch teuer. Die Elementanalytik war einer der Protagonisten. Der Chronist und
seine Mitarbeiterinnen und Mitarbeiter sind stolz, diese Entwicklung ein Stück
begleitet zu haben.
Schlusssatz
Der erste Naturforscher, der Metalle und Mineralstoffe analytisch beackerte, soll Georg Agricola (1494-1555) gewesen sein. Wenn das kein Omen war? (agricola (lat.): Bauer, Landwirt)
Literatur
Dem
Originalbeitrag in der o.g. Festschrift ist ein umfangreiches
Literaturverzeichnis angefügt. Auf Anfrage an eine der auf der Startseite
angegebenen Mailadressen des Autors kann es angefordert werden.
![]()